-
Basecalling overview
A user can basecall and demultiplex their data directly in MinKNOW after a sequencing experiment has finished to avoid having to use command-line tools, or to re-analyse old data using the latest basecalling models. Reads can also be aligned against a reference post-sequencing.
Note: Both barcoding and alignment can be run on FASTQ, POD5 or FAST5 reads when coupled with basecalling.
-
Click 'Analysis' on the start page.
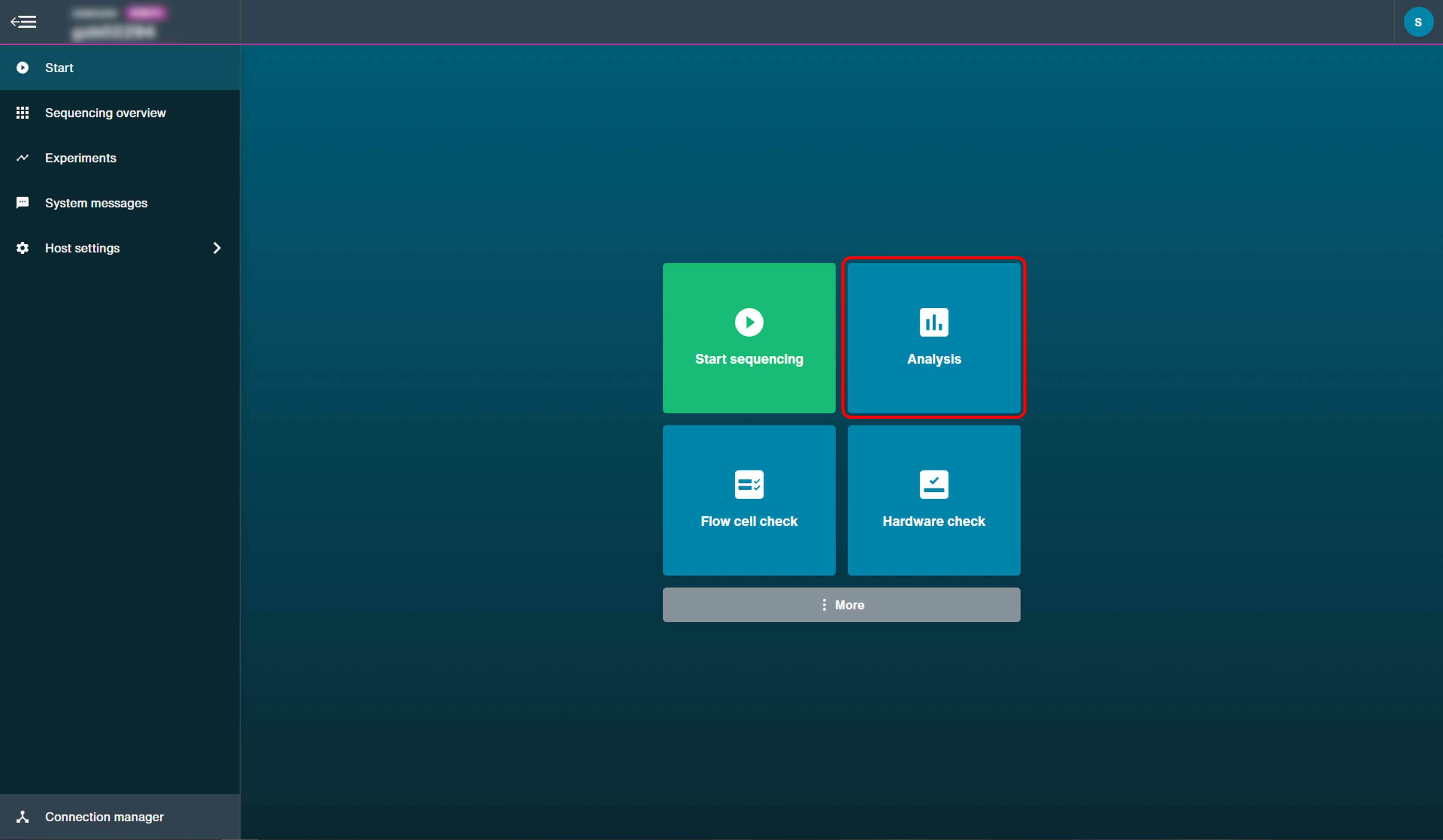
-
Click 'Basecalling' to open the post-run basecalling set-up options.
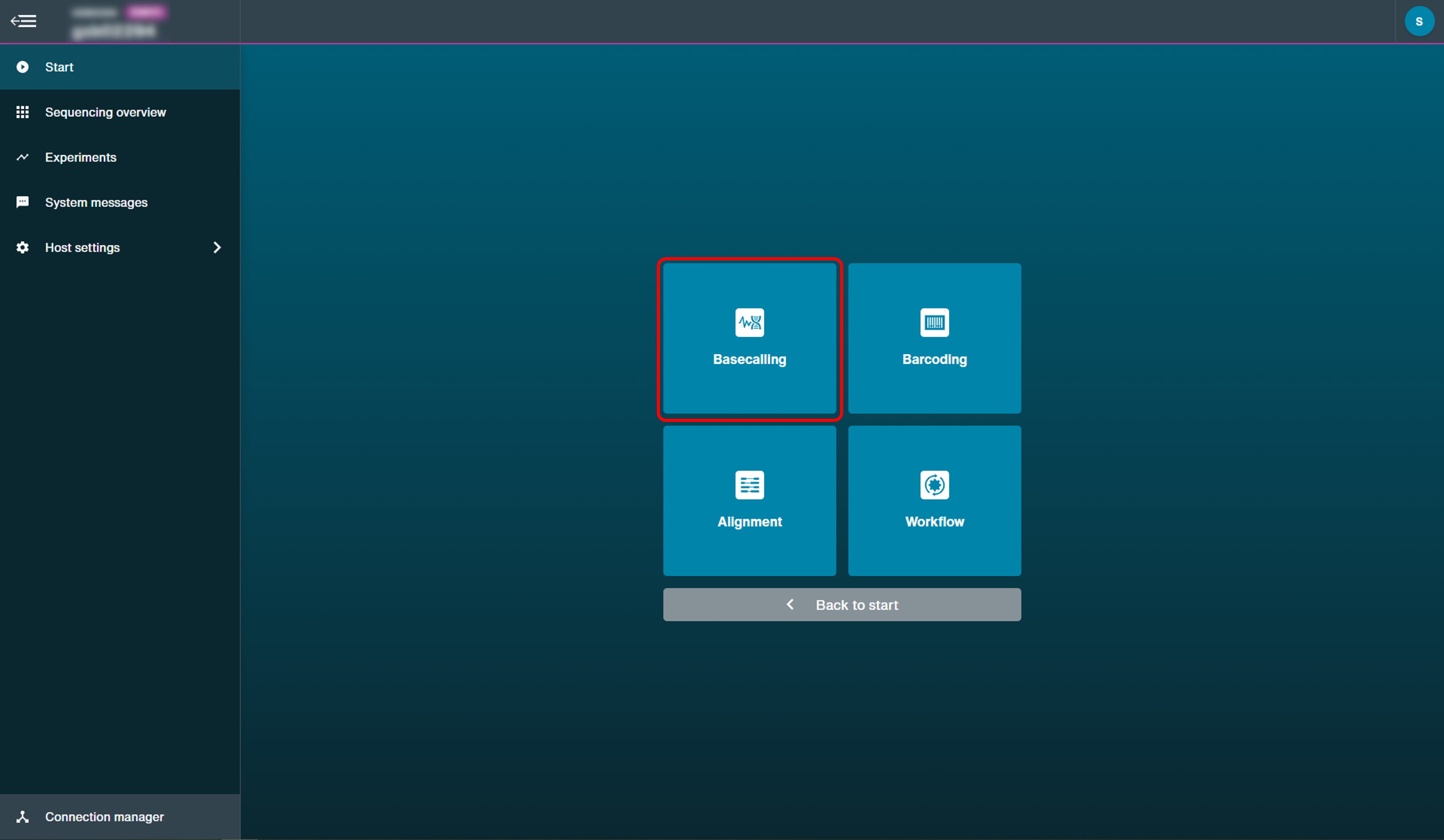
-
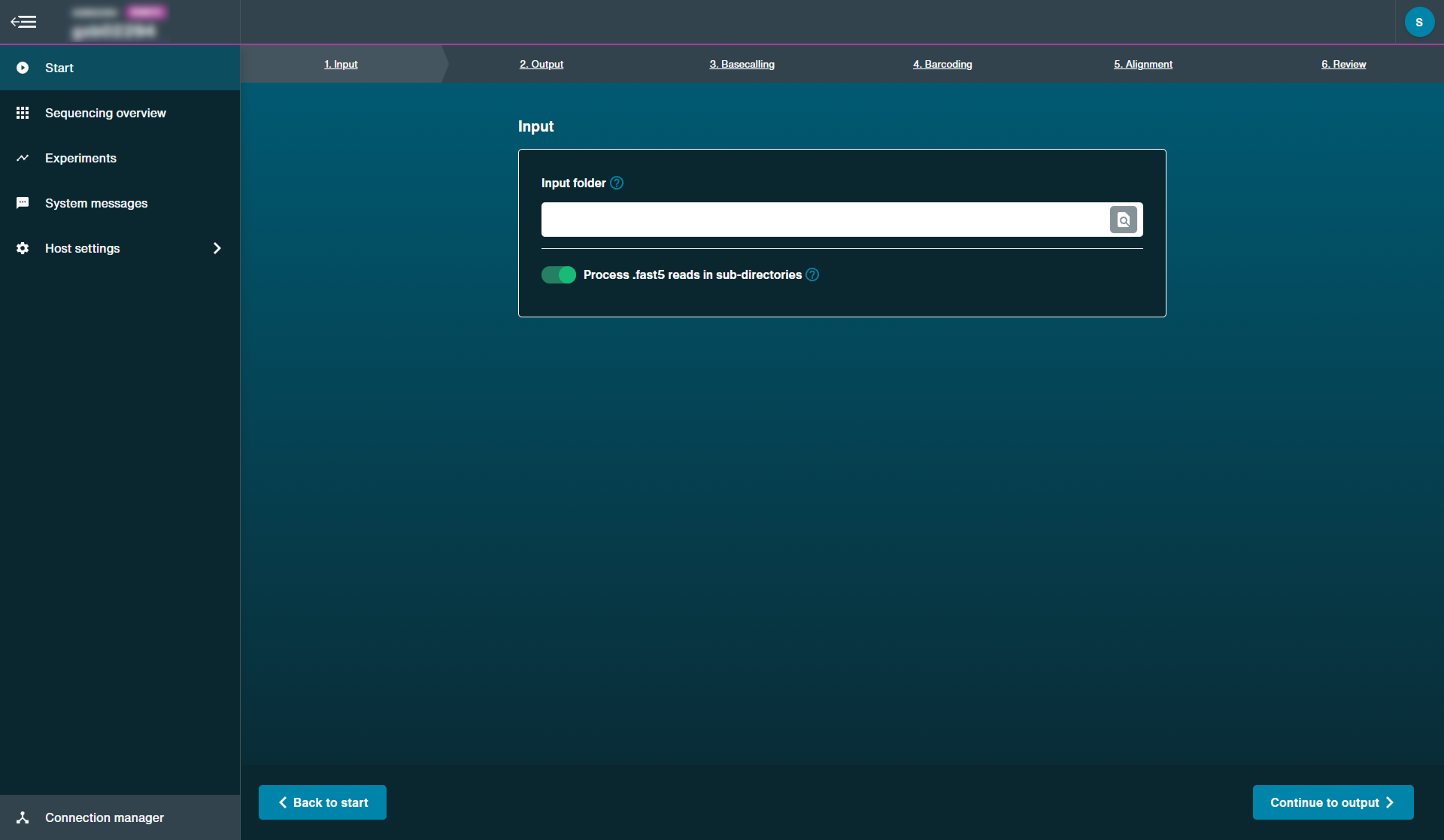
Select input folder containing the FAST5 files to basecall from a previous run.
Select whether you want to process FAST5 reads in sub-directories.
If your chosen read input folder contains sub-directories with FAST5 files, you can choose whether to basecall FAST5 files in these sub-directories by enabling this option.
-
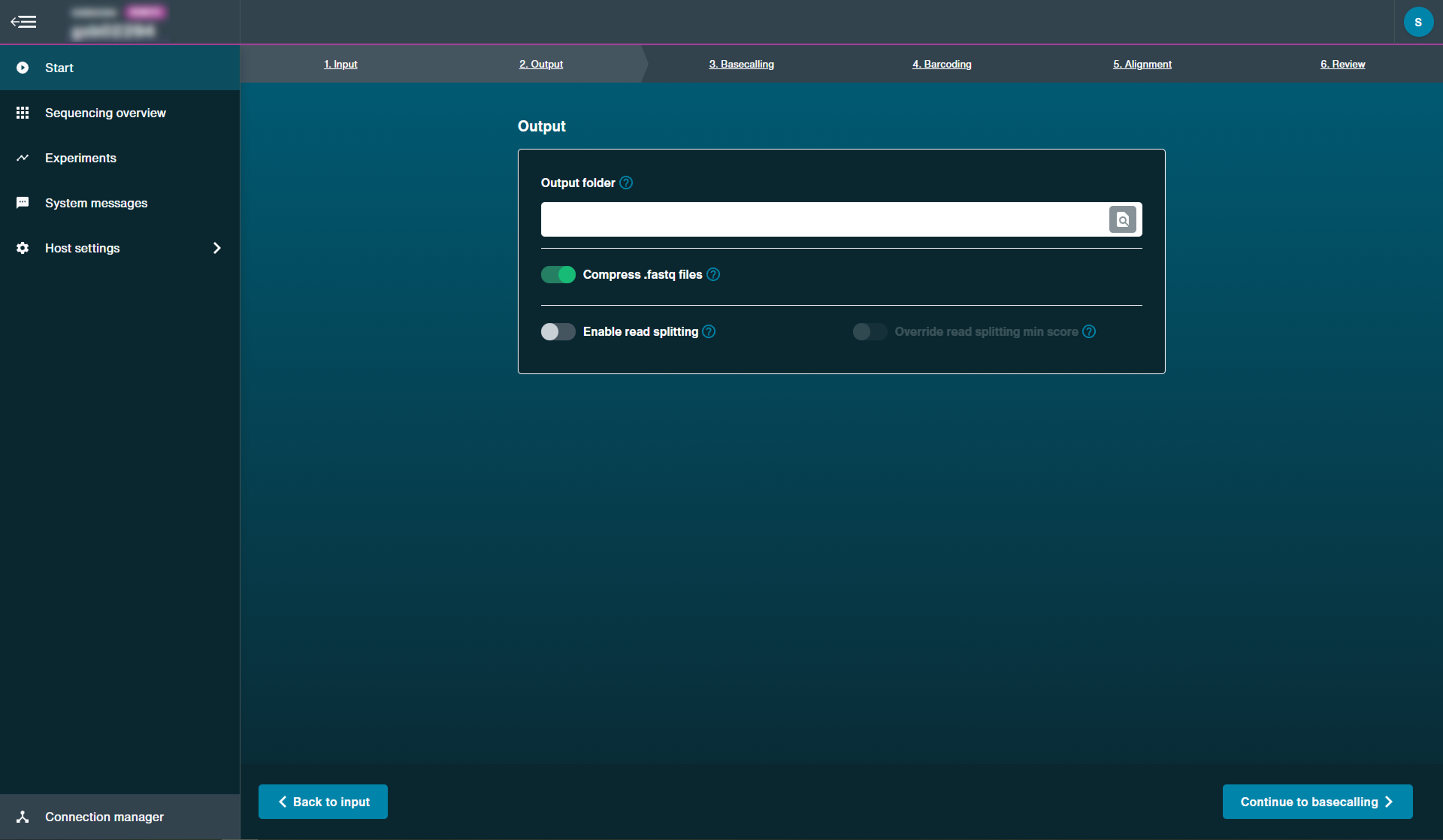
Select output folder and file type.
By default, MinKNOW will create a
/basecalledfolder in/data. You can set a different folder in which to save the basecalled reads.
Note: This must be a sub-folder of/datadirectory.Select whether to output FAST5 files. If selected, these files will be written to a folder within
/basecalledcalled/workspace, if selected.Users are also able to enable read splitting options post-run.
-
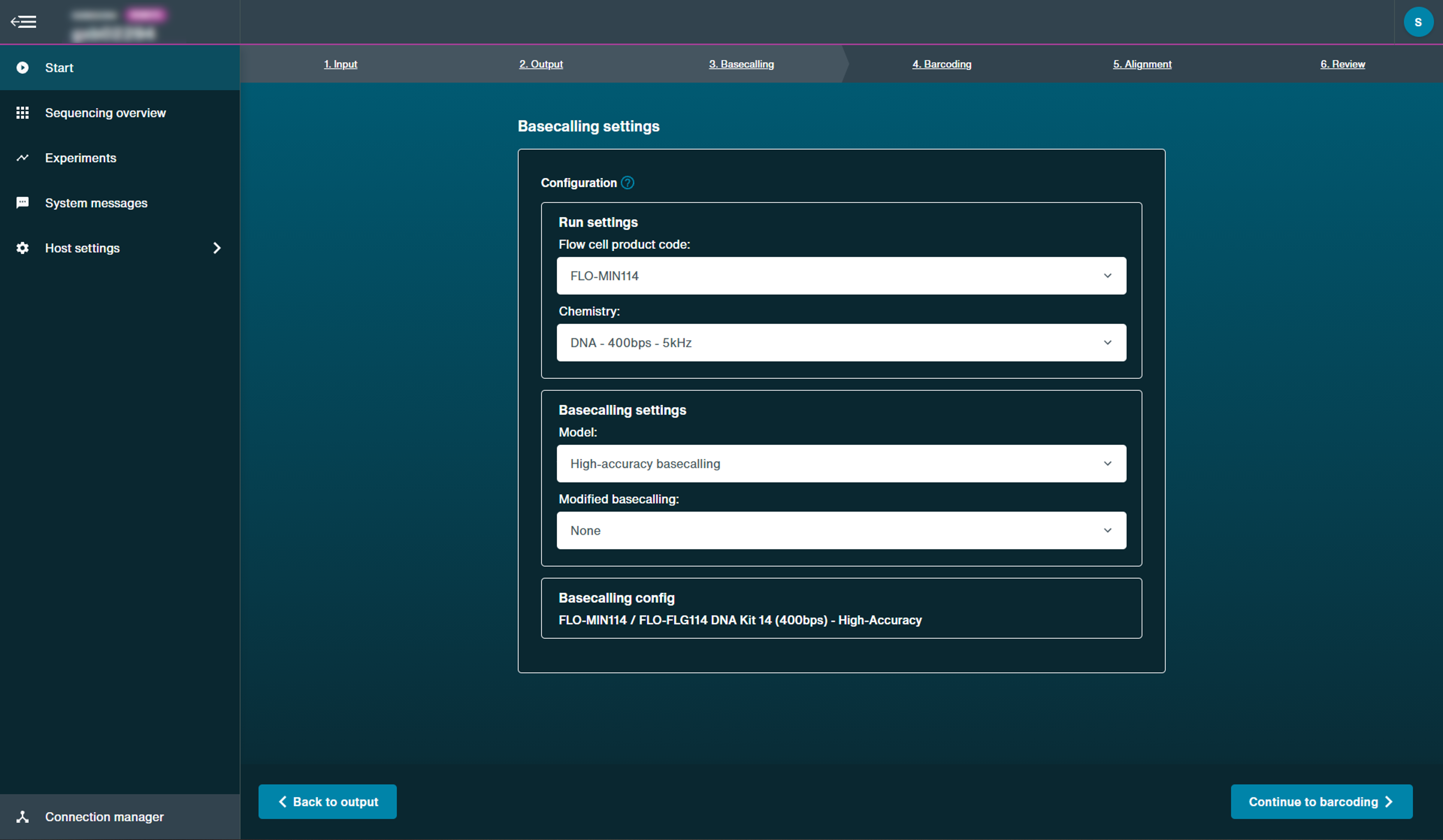
Choose basecalling model from the drop-down menu.
Set-up the configuration of your basecalling run using the drop-down menus:
- In the run setting, select the parameters used to initially sequence the data.
- In the basecalling settings, select the basecall model used to process raw signal, and whether to use modified basecalling.
Once all the parameters have been selected, the basecalling configuration will be displayed in the lower panel.
Given the recent improvements to basecalling accuracy, Q-score cut-off for reads put into the "pass" folder has been increased to:
Fast basecalling model >8
High accuracy (HAC) model >9
Super accurate (SUP) model >10 -
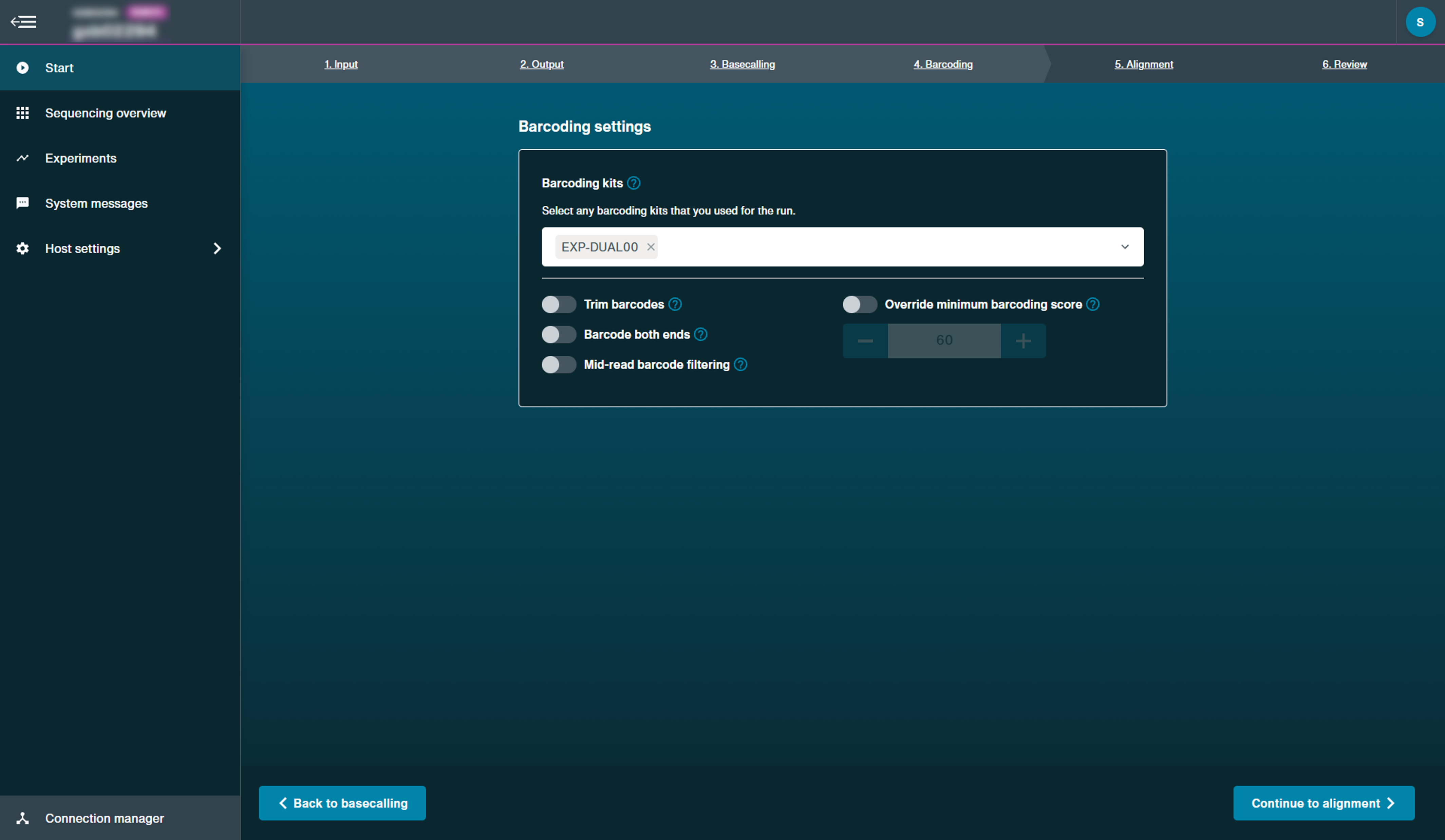
Optional actionSelect any barcoding kits used for the run from the drop-down menu.
MinKNOW will write out basecalled reads into barcode-specific folders and enable demultiplexing during basecalling.
Users are able to choose specific settings, including trimming barcodes and minimum barcoding scores.
-
Optional actionSelect to use alignment and import a reference sequence in a FASTA file.
We currently only recommend uploading an alignment reference locally for bacterial-sized genomes. Upload can take a few minutes and is compute dependent.
A reference file must by uploaded as a FASTA file that can contain multiple entries in the same file (e.g. multiple chromosomes).
However, a .bed file may also be uploaded alongside the reference FASTA file. The BED file option can be used when the user is interested in a particular region of the reference (e.g. specific gene in the chromosome).
Alignment generates FASTQ output files when performed with basecalling.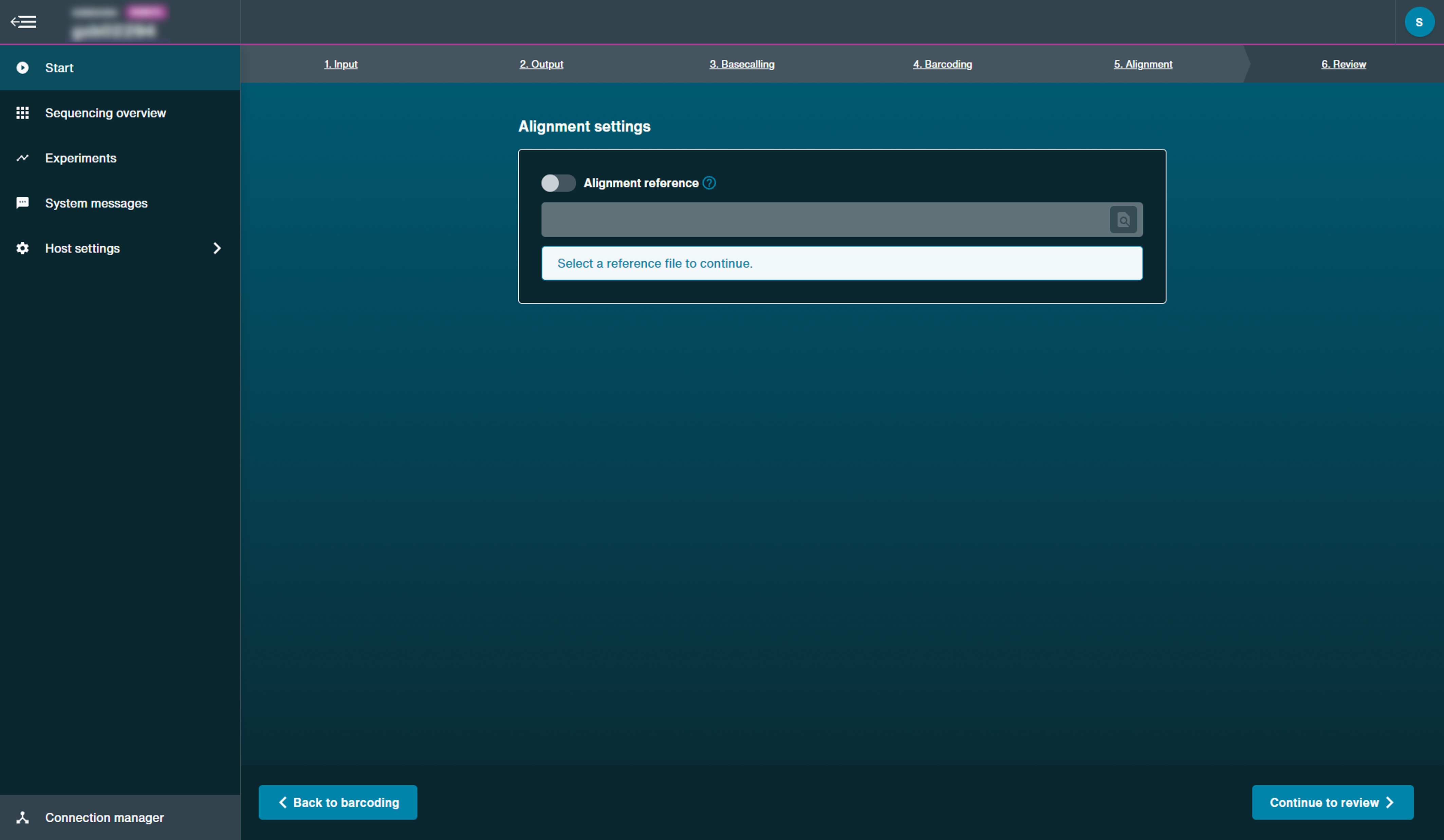
-
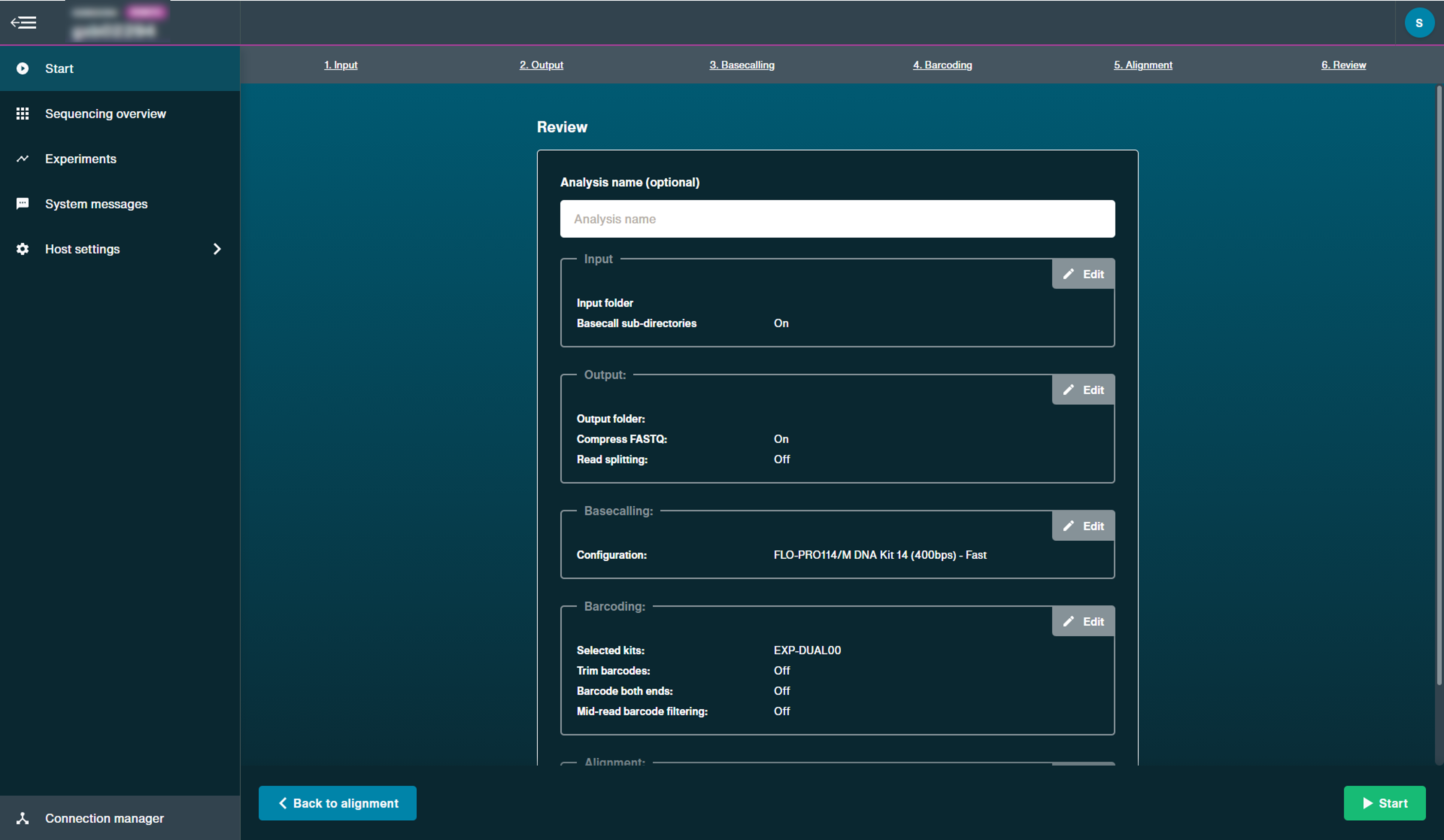
Click 'Start' to begin post-run basecalling.