-
How to start sequencing
The sequencing device control, data acquisition and real-time basecalling are carried out by the MinKNOW software.
We recommend basecalling with the high accuracy (HAC) basecaller in real-time with BAM selected as output type using the P2 Solo or P24/P48 device.
A BAM file must be generated as this is required as input into the wf-human-variation workflow. We recommend aligning the BAM output after live basecalling to prevent slowing sytem processing or to provide a reference genome along with the unaligned BAM file in the wf-human-variation workflow. Using mapped BAM as input, the workflow will take 1-2 hours. If unmapped BAM is used as input, the workflow will take approximately 5-8 hours. Further information is available in Downstream analysis.
Refer to the links below for detailed instructions for setting up the device and sequencing run:
PromethION 24 and 48: "Starting a sequencing run with PromethION 24 and 48"
PromethION 2 Solo: "Starting a sequencing run on PromethION 2 Solo"
Below are the recommended sequencing parameters for MinKNOW.
-
MinKNOW settings for 10kb human sample on PromethION
We recommend using the modified bases option for basecalling and ensuring a BAM output is selected when setting up your MinKNOW run. The remaining sequencing parameters are kept to default.
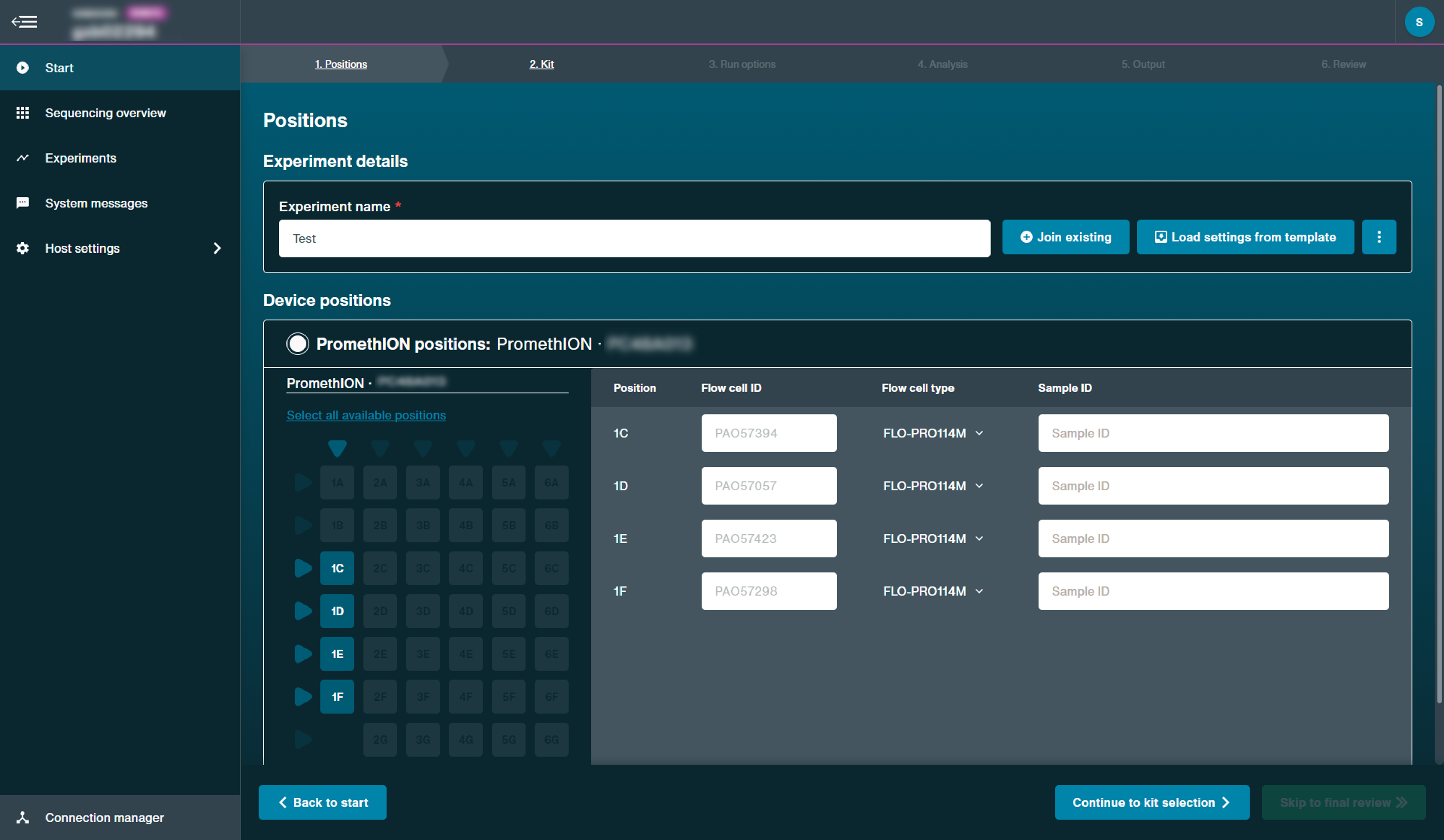
Below are our current recommendations:Positions
Flow cell position: [user defined]
Experiment name: [user defined]
Flow cell type: FLO-PRO114M
Sample ID: [user defined]
Kit
Kit selection: Ligation Sequencing Kit (SQK-LSK114)

Run options
Run limit: 72 hours [default]
Minimum read length: 200 bp [default]
Adaptive sampling
Enrich or deplete sequening: Off [default]
Barcoding balance: Off [default]Advanced options
Active channel selection: On [default]
Time between pore scans: 1.5 [default]
Reserve pores: On [default]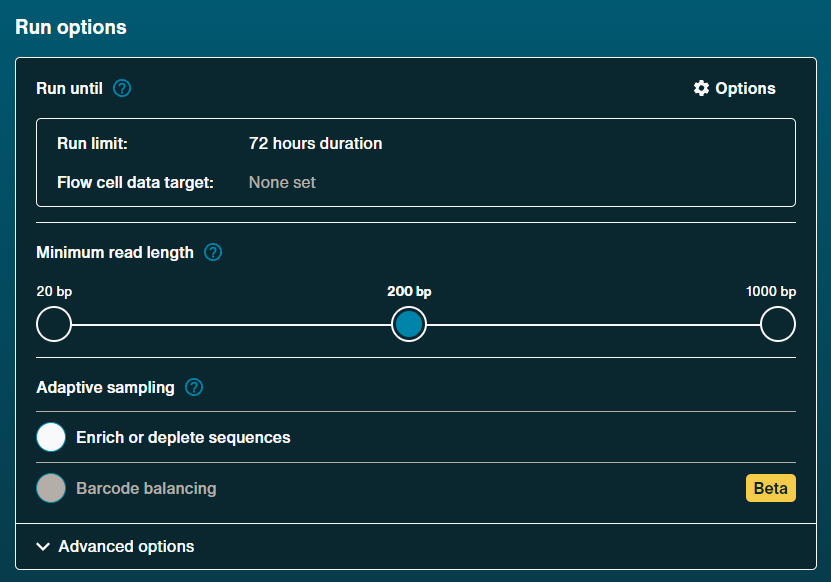
Analysis
Basecalling
Basecalling: On [default]
Modified bases: On with '5mC' selected
Model: High-accuracy basecalling (HAC) [default]Barcoding
Barcoding: Disabled [default]Alignment
Reference sequence: Off [default]Note: We do not currently recommend live alignment during sequencing, as it can slow down system processing.

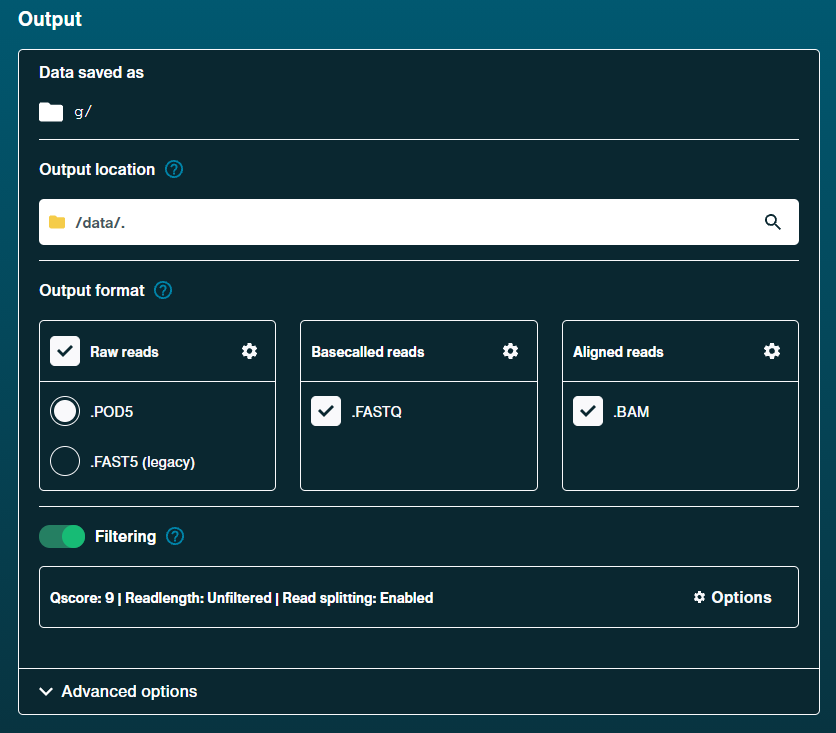
Output
Output format
.POD5: On [default]
.FASTQ: On [default]
.BAM: OnFiltering: On [default]
Qscore: 9 [default]
Readlenth: Unfiltered [default]
Read splitting: Enabled [default]