-
How to start sequencing duplex data
The sequencing device control, data acquisition and real-time simplex basecalling are carried out by the MinKNOW software. After simplex data has been basecalled, duplex basecalling is performed on Dorado, which is detailed below the MinKNOW settings.
Ensure you are using the most recent software version of MinKNOW.
We recommend basecalling with the Fast basecaller in real-time using the PromethION 24 or 48 device. After simplex basecalling on MinKnow, the data can be rebasecalled using the super accurate (SUP) model when performing duplex basecalling on Dorado.
For detailed instructions for setting up the device and sequencing run, see the MinKNOW protocol section for "Starting a sequencing run with PromethION 24 and 48".
For further detailed instructions for simplex and duplex basecalling, see the Kit 14 sequencing and duplex basecalling document.
-
MinKNOW settings for the Duplex experiment
For the Duplex experiment, we recommend keeping the sequencing parameters to their default settings for simplex basecalling.
Below are the current recommendations:
Positions
Flow cell position: [User defined]
Experiment name: [User defined]
Flow cell type: FLO-PRO114M
Sample ID: [User defined]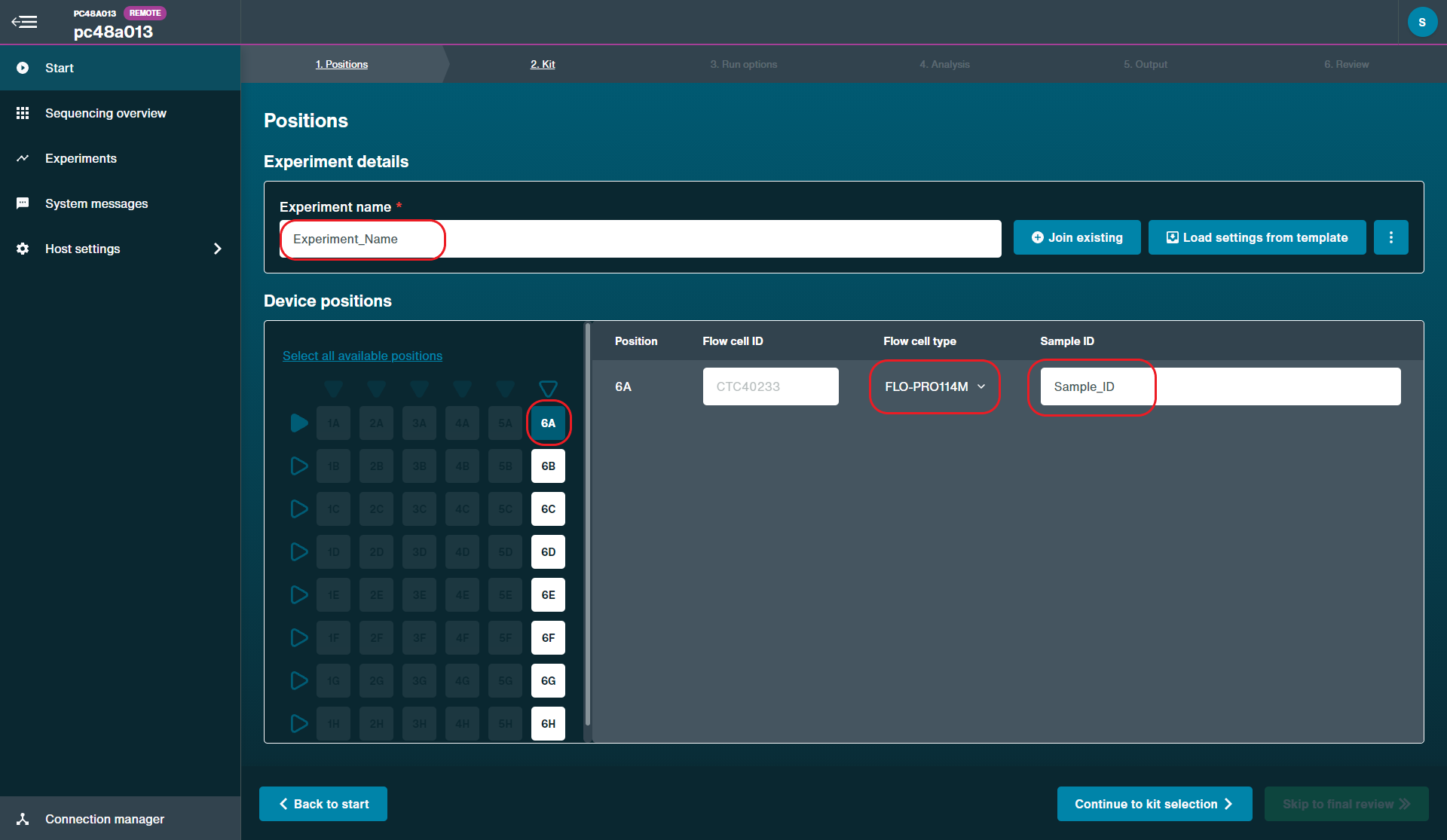
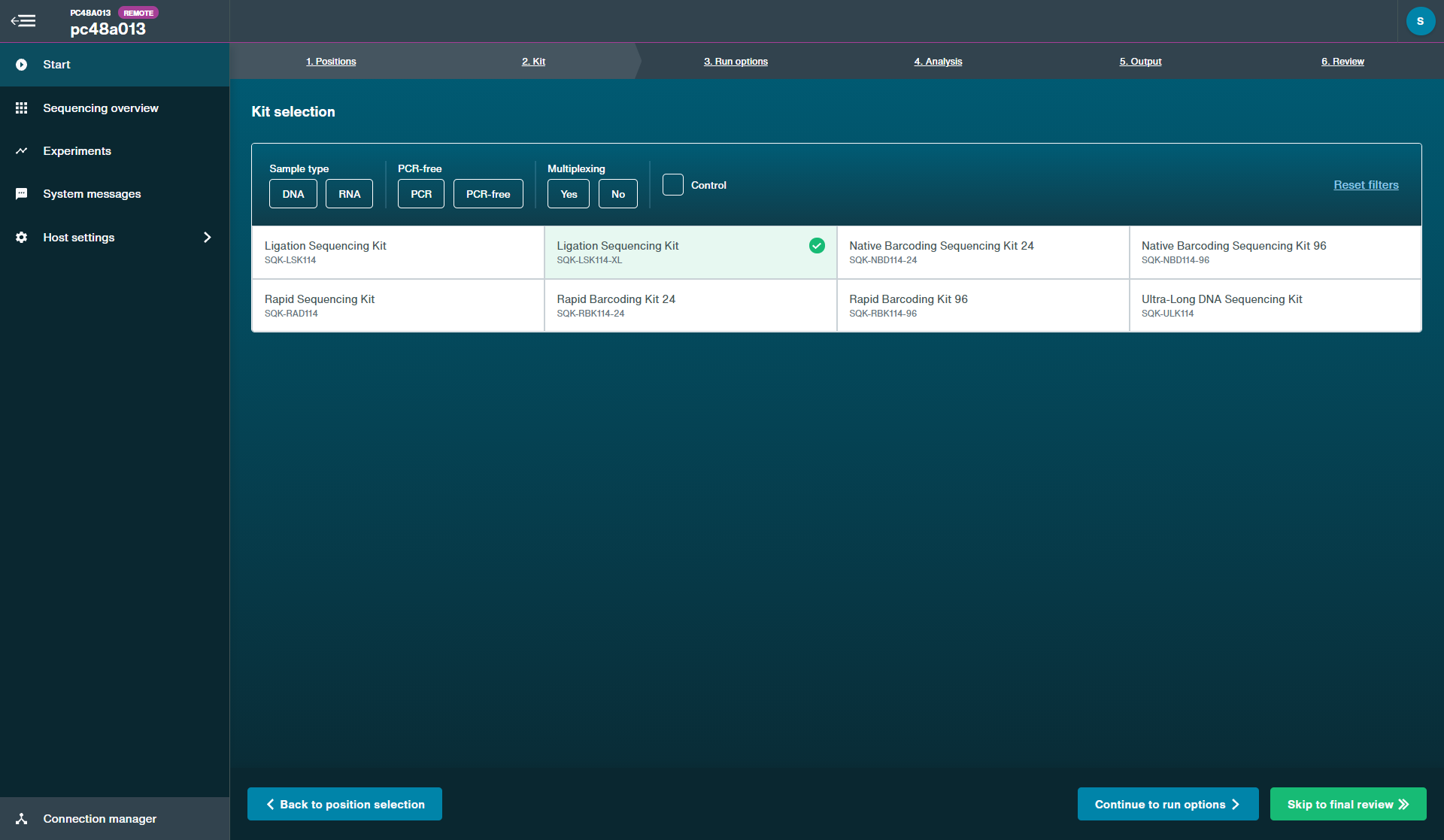
Kit selection:
Kit selection: Ligation Sequencing Kit XL V14 (SQK-LSK114-XL)
Run options:
Run duration: 100 hours
Minimum read length: 200 bpAdaptive sampling
Enrich or deplete sequencing: Off
Barcoding balance: Not applicableAdvanced options
Active channel selection: On
Time between pore scans: 1.5 hours
Reserve pores: On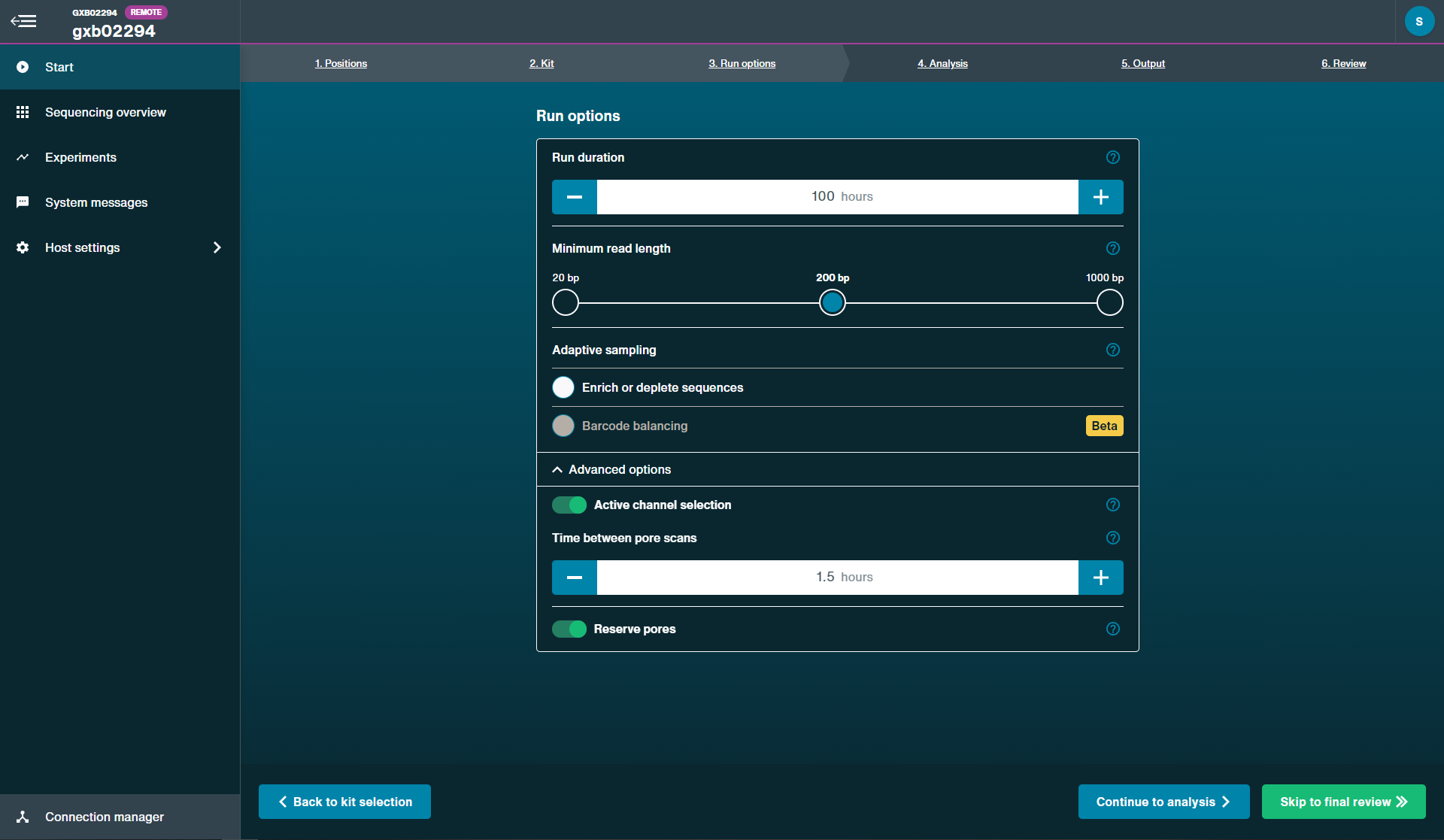
Analysis:
Basecalling
Basecalling: On
Modified bases: On with '5mC' selected
Model: Fast basecallingBarcoding
Barcoding: Disabled
Alignment
Reference sequence: Off
Note: We do not currently recommend live alignment during sequencing as it can slow down system processing.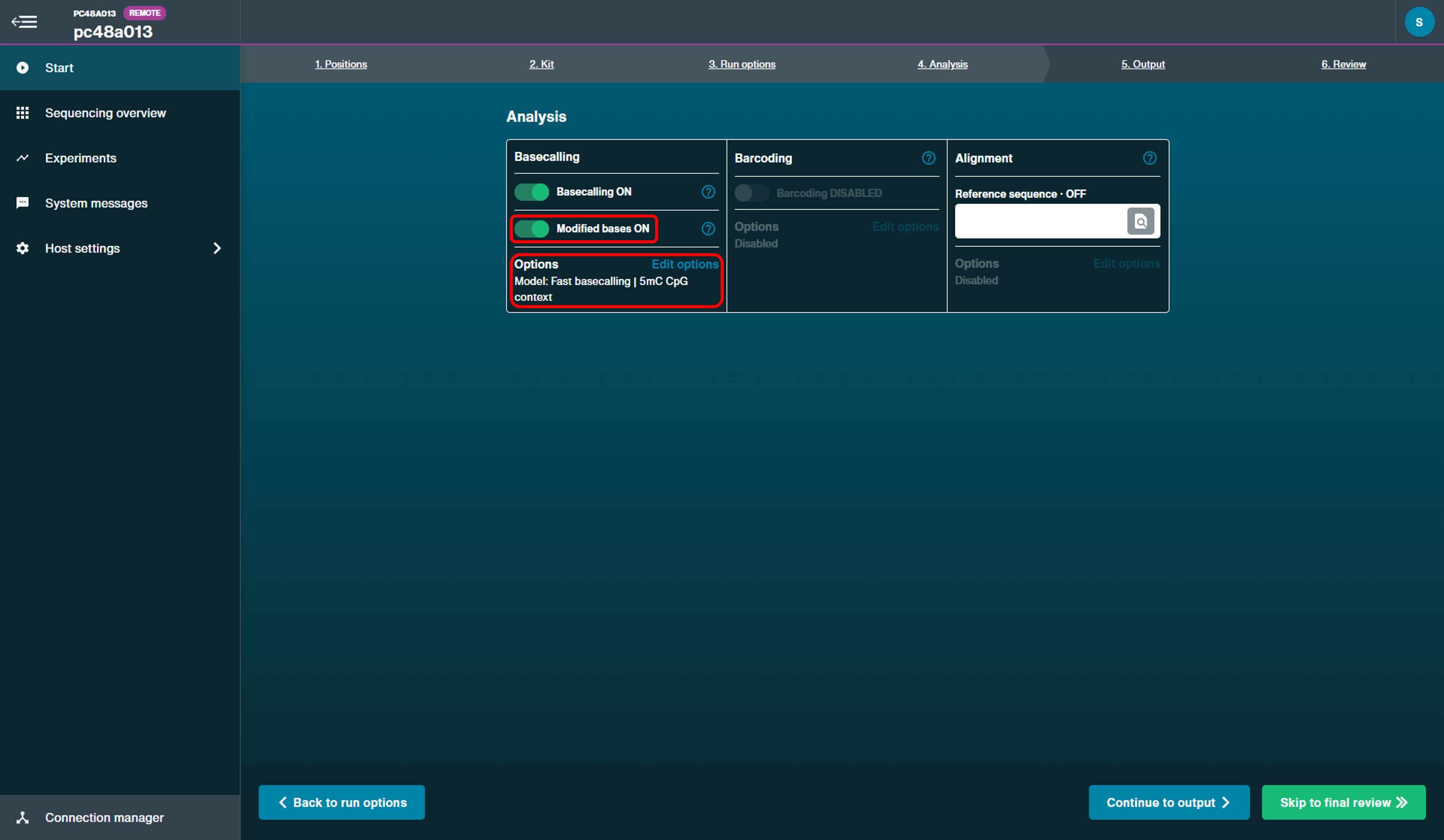
Output:
Output format
Raw reads: On
.FAST5: Off
.POD5: On
.FASTQ: On
.BAM: On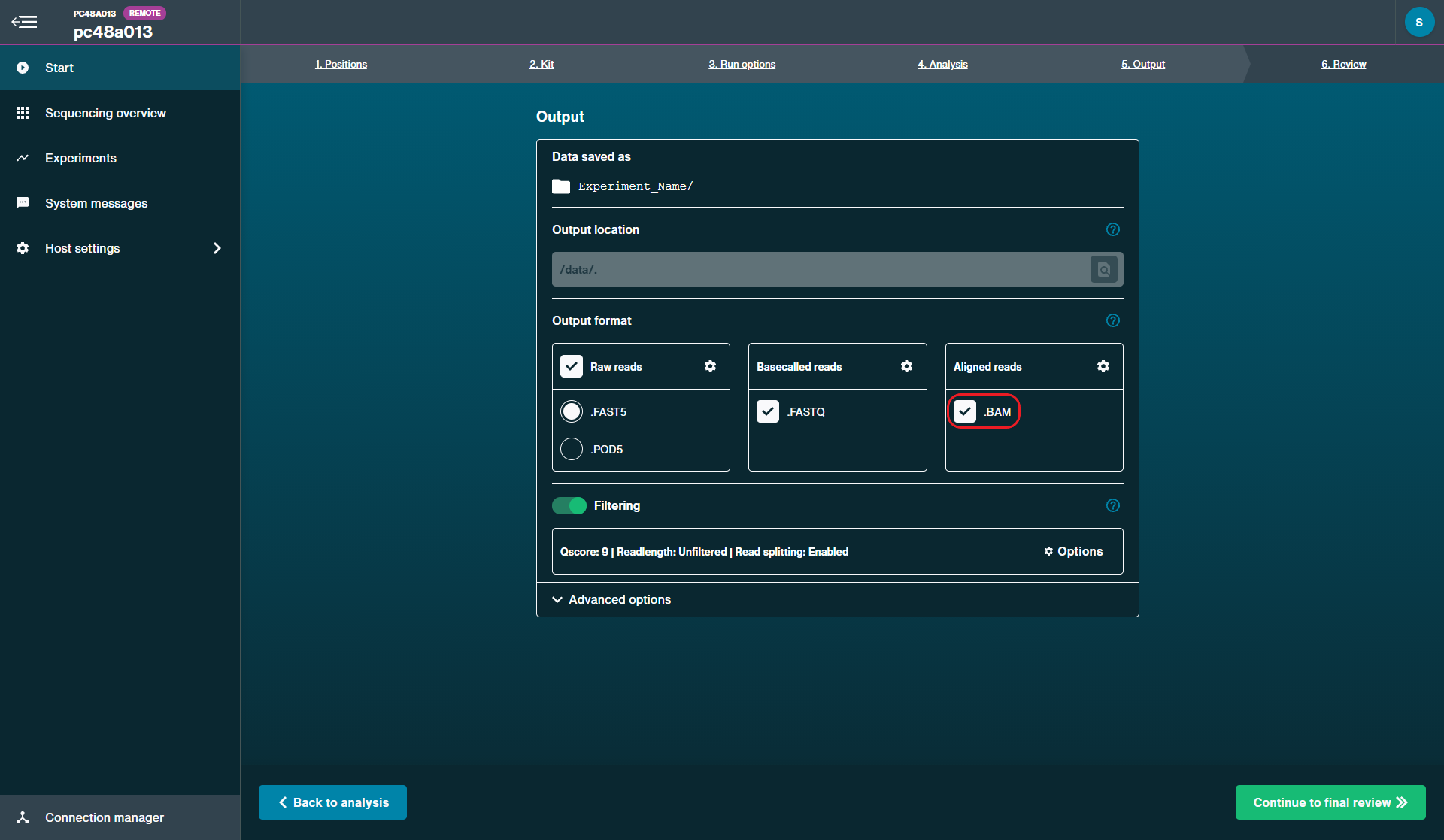
Filtering: On
Qscore: 8
Read length: Unfiltered
Read splitting: Enabled
Override read splitting min score: On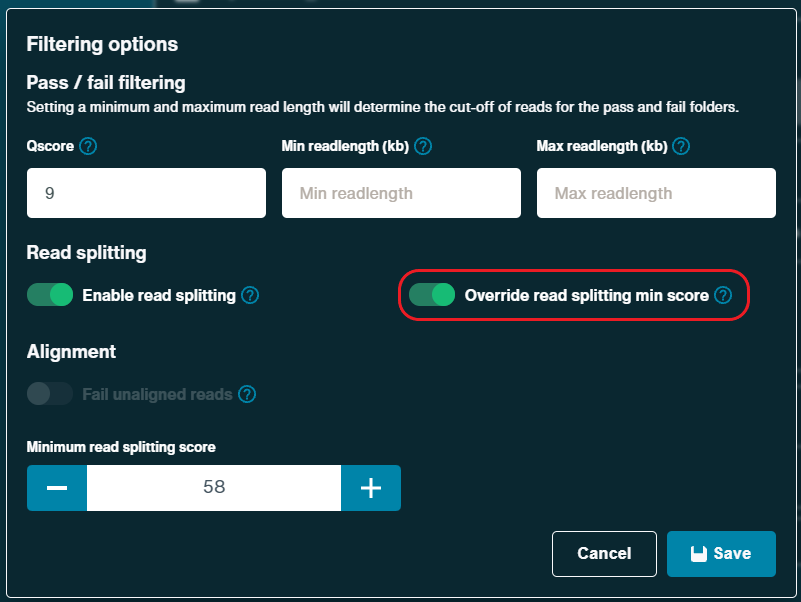
-
Overview of performing duplex basecalling:
Set up sequencing parameters in MinKNOW to perform simplex basecalling as described in "Basecalling Kit 14 simplex data".
a. Basecall using the Fast basecaller.
b. Output .POD5 files.Using Dorado, re-basecall your simplex data with the following command to output simplex and duplex reads using the super-accurate (SUP) basecalling model. Other basecalling models can be used, as listed on the Dorado Github page.
$ dorado duplex dna_r10.4.1_e8.2_400bps_sup@v4.1.0 pod5s/ > duplex.bamDorado is a high-performance basecaller which is used to perform duplex basecalling. For further information about Dorado, please see the Dorado Github page.
Note: When running Dorado, we recommend stopping other basecalling for the best performance by maximising memory available to Dorado. This can be stopped and restarted when Dorado has finished via the GUI on MinKNOW.
Guppy may also be used to duplex basecall.





