-
Alignment overview
A user can align their data directly in MinKNOW after a sequencing experiment has finished, using a FASTQ file from a previous run.
The reference file of bacterial-sized genomes must be uploaded locally as a FASTA file. Alignment hits from these files are used to populate the alignment graphs.
A BED file may also be uploaded alongside the reference FASTA file when the user is interested in a particular region of the reference (e.g. specific gene in the chromosome). The BED file alignment hits will be highlighted in the sequencing .txt file generated in the data folder.
When alignment is run independently from basecalling, BAM files are generated.
-
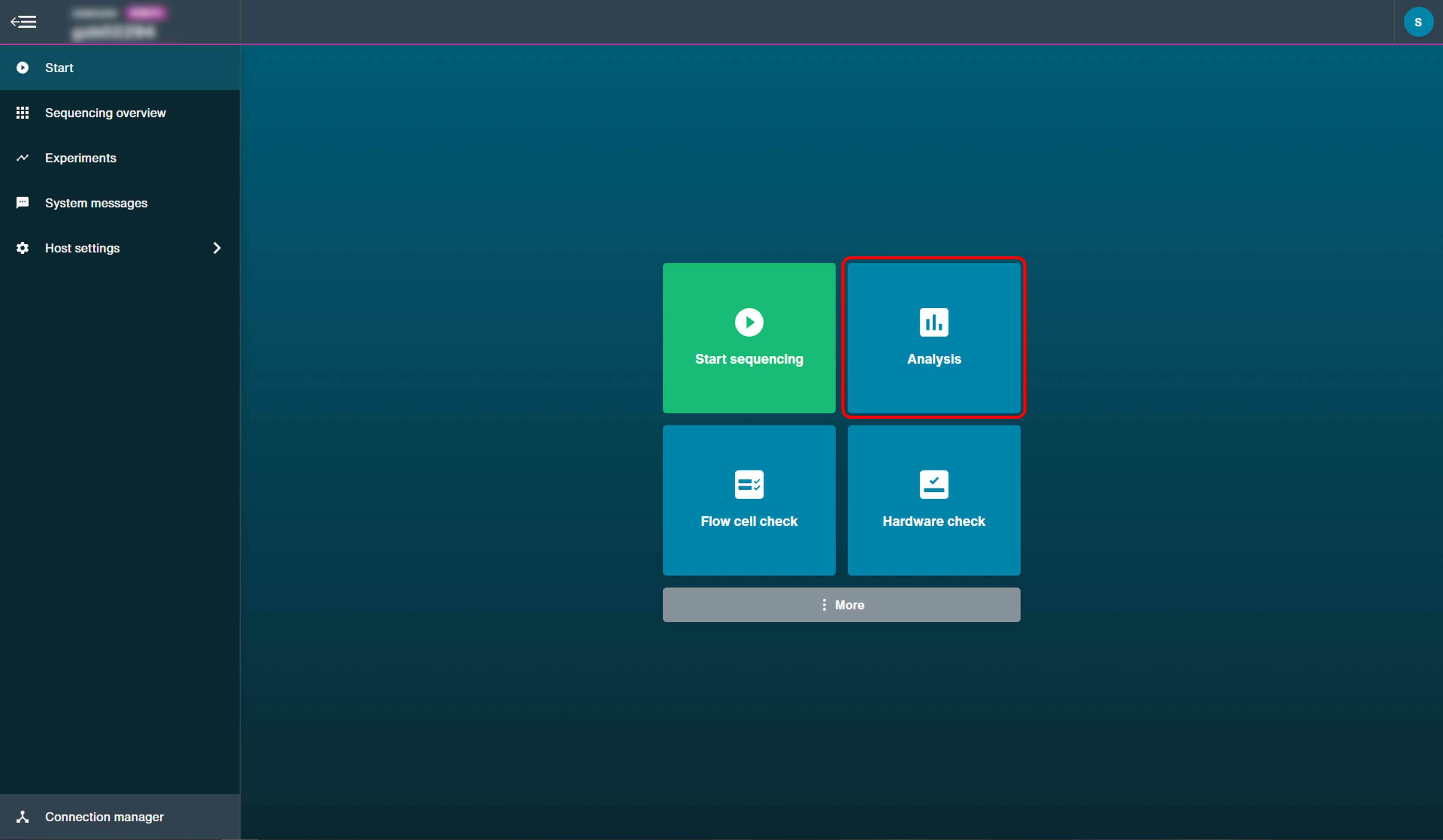
Click 'Analysis' on the start page.
-
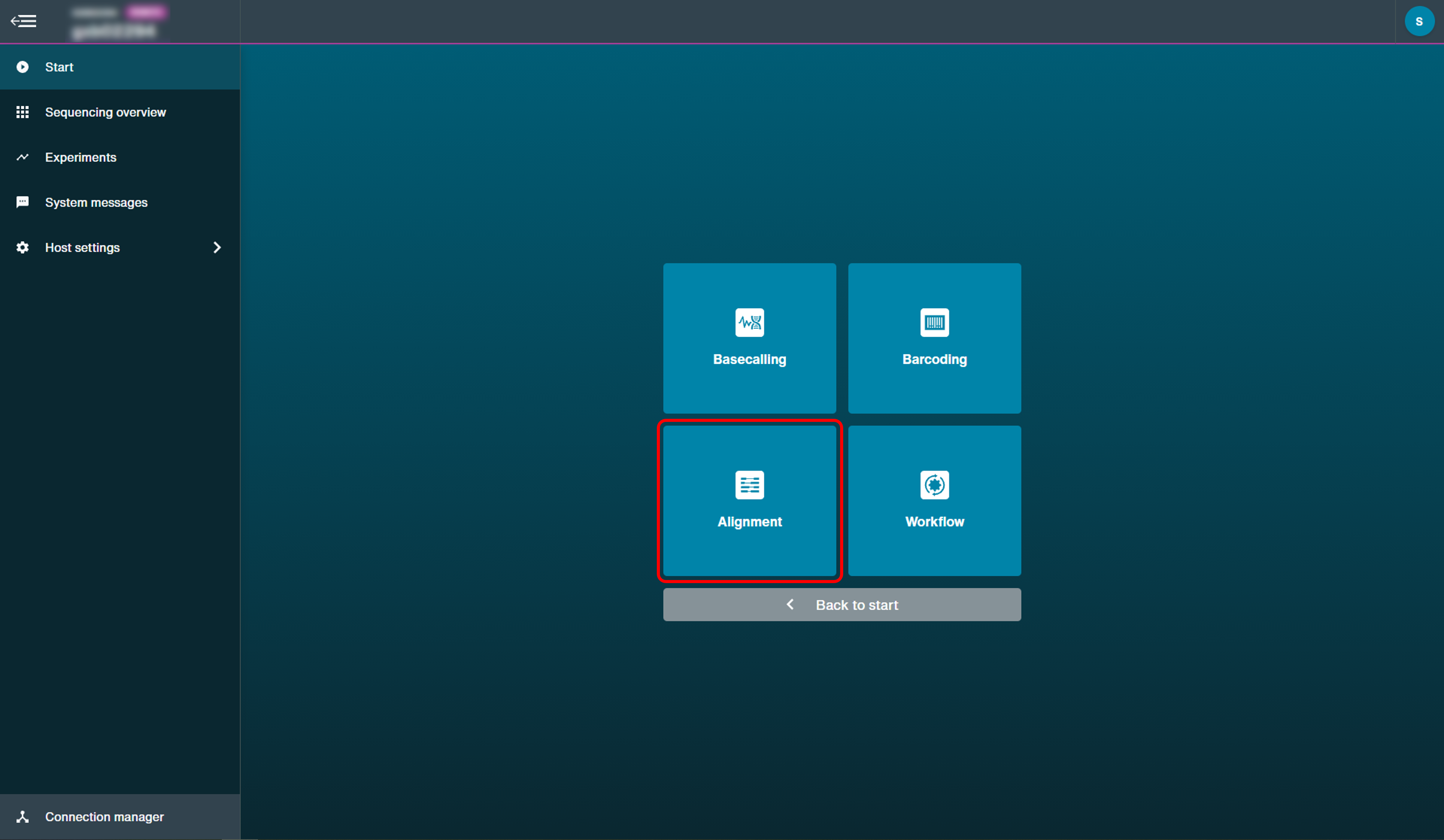
Click 'Alignment' to open the post-run analysis set-up options.
-
Select the input file containing the FASTQ data to carry out alignment from a previous run.
Select whether you want to process FASTQ reads in sub-directories.
If your chosen read input folder contains sub-directories with FASTQ files, you can choose whether or not to align FASTQ files in these sub-directories by switching this option on.
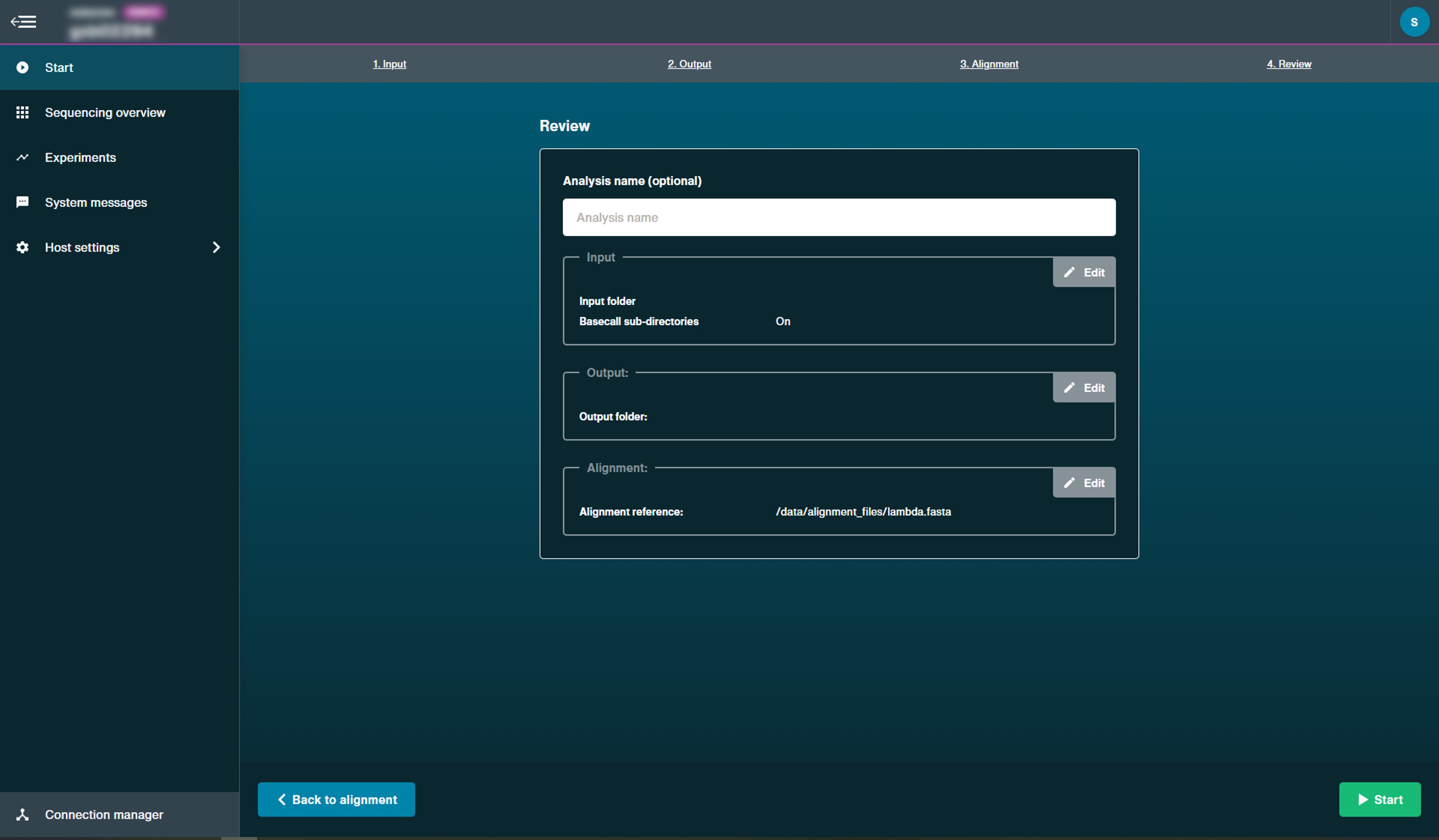
Note: Data can only be saved to the
/datafolder.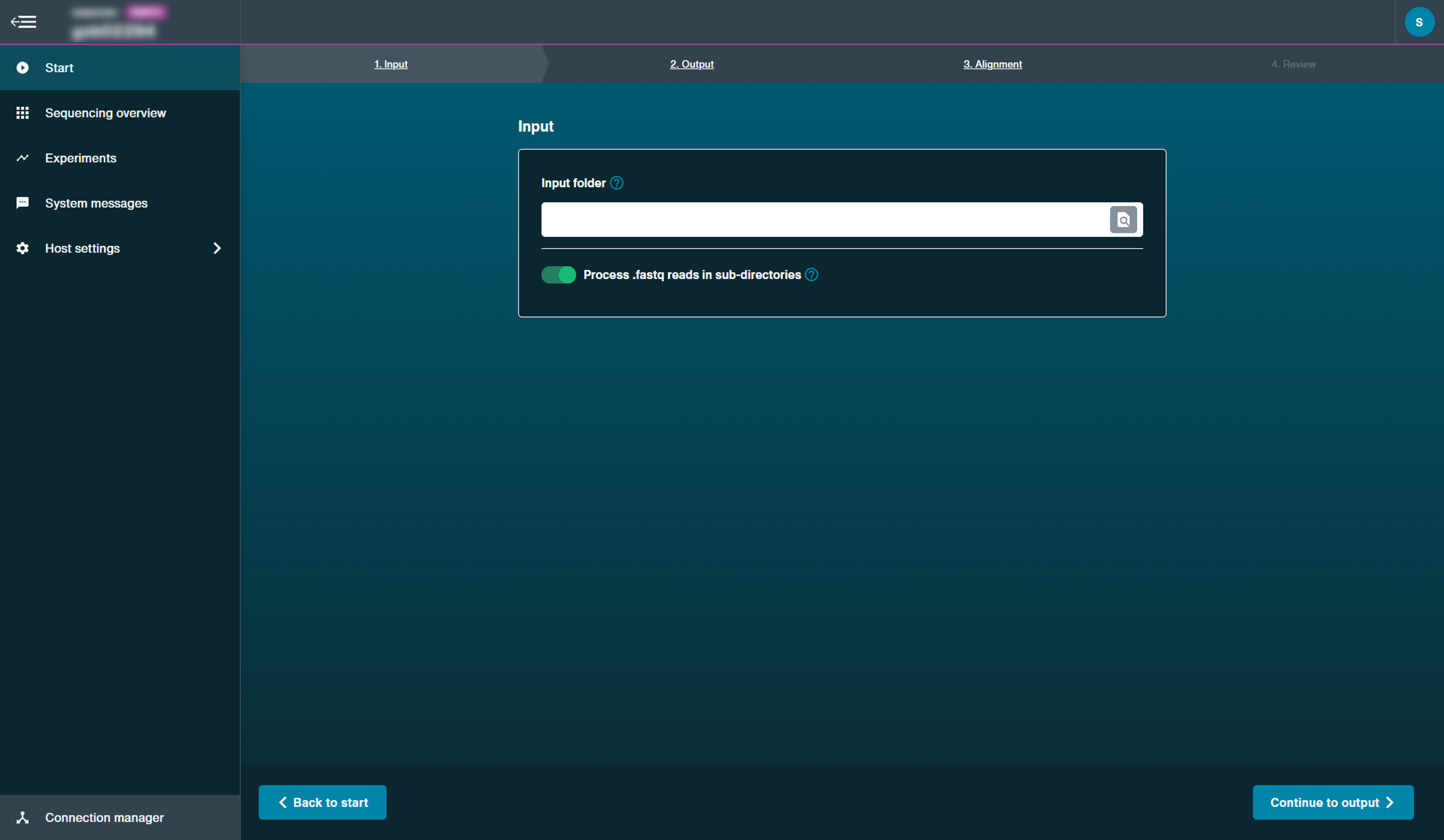
-
Choose the output folder for the post-run aligned data.
By default, MinKNOW will create an
/alignment/folder in/data/. You can set a different folder which to save the aligned reads.
Note: This must be a sub-folder of/datadirectory.
-
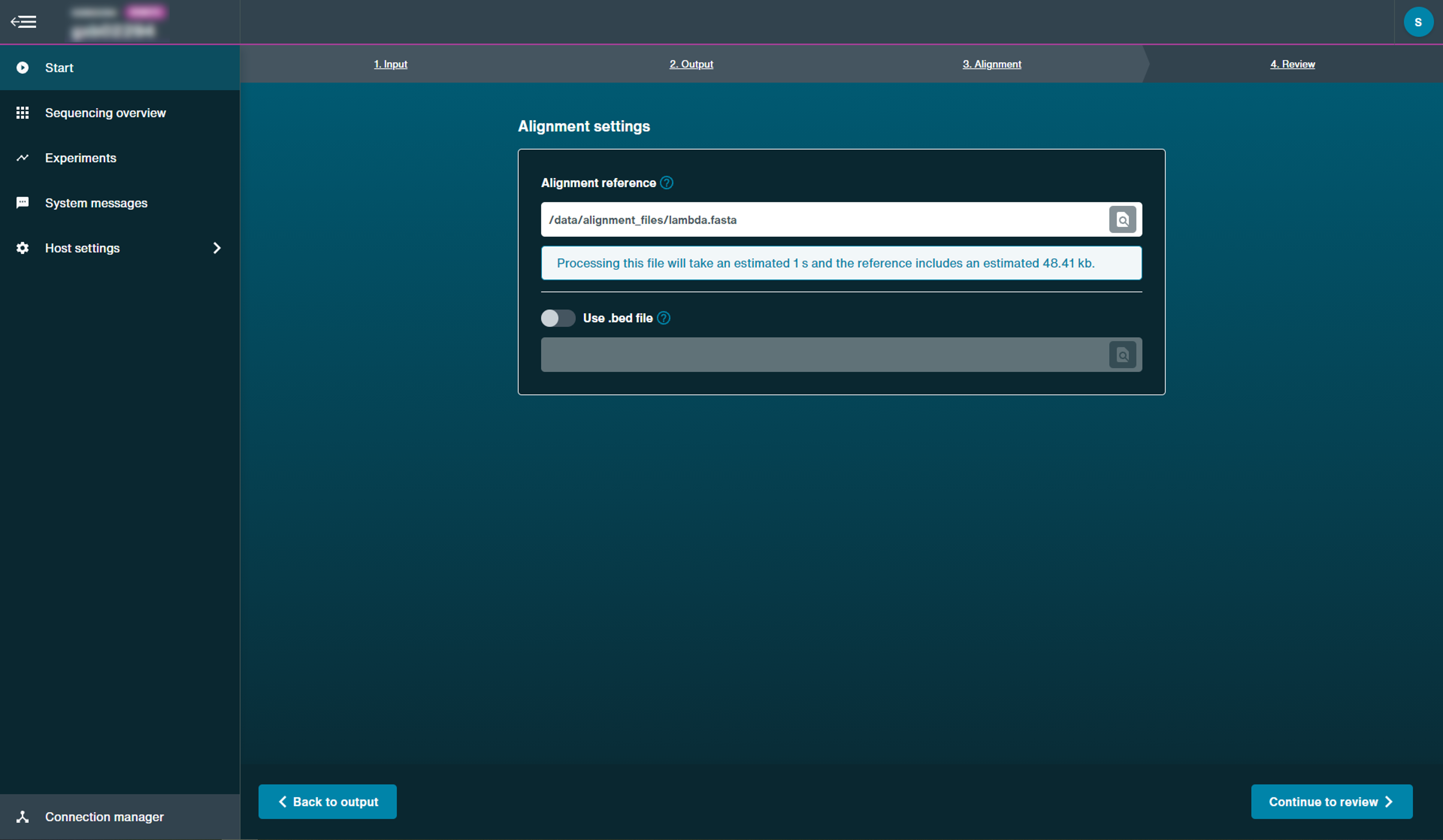
Select the alignment reference to carry out the post-run alignment.
The alignment reference must either be a FASTA file.
However, there is also the option to use a BED file alignment reference as well.
-
Click 'Start' to begin post-run alignment.