-
For GridION and PromethION experiments, or when multiple MinION Mk1Bs or Mk1Cs are running simultaneously, it may be preferred to upload sample names and corresponding flow cell positions from a CSV file, rather than manually.
The sample sheet may describe flow cells being run at one or more than one position. The columns available are as follows, though some are optional:
Column title Description Notes flow_cell_id Defines the flow cell ID which applies to the sample sheet row. Used to identify which positions to apply values to in MinKNOW.
This is optional if position_id is specifiedposition_id Defines the flow cell position which applies to the sample sheet row. Used to identify which position to apply values to in MinKNOW.
This is optional if flow_cell_id is specified.sample_id Defines the sample ID to be applied in the run An individual position can only have one sample_id assigned to it when starting a run.
This is optional.experiment_id Defines the experiment ID to be applied Each row in the sample sheet must contain the same experiment_id value for the sample sheet to only have a single experiment_id value defined overall. All entries in each row will be validated. flow_cell_product_code Defines the product code of the flow cell Used to find the correct sequencing script to start the run kit Defines the kit and any expansion kits used with the sample Used to find the correct sequencing script to start the run. The sample sheet must contain only one sequencing kit, but expansion kits are not limited. If expansion kits are additionally defined, they should be separated by a space: e.g. SQK-LSK109 EXP-NBD104 EXP-NBD114
Note: Ensure the correct format of the kit name is used, converting any full stops in the kit name to a dash. For example, the Rapid Barcoding Kit V14 96 (SQK-RBK114.96) format in the sample sheet would be SQK-RBK114-96.For experiments that involve barcoding, an alias is associated with each barcode (or pair of barcodes where appropriate). Additional columns are available for barcoding runs:
Column title Description Notes alias User-specified string which applies a given label to a specific barcode or barcode pair. The alias cannot be an existing barcode folder name e.g. 'unclassified', 'classified' and 'mixed' are not allowed. The alias must be between 1 and 40 characters. The character set needs to match the following format: /^([0-9a-zA-Z-_]and only contain the following characters: numbers, upper/lower-case letters, dashes, and underscores.type One of the options: test_sample, positive_control, negative_control, no_template_control This is optional. One or two columns define the barcode or barcode pair. The exact form is dependent on whether single or dual barcoding is being used.
Single barcoding: the sample sheet contains one barcoding arrangement column:
Column title Description Notes barcode The barcode identifier for the row e.g. barcode01 Dual barcoding: the sample sheet contains two barcoding arrangement columns:
Column title Description Notes internal_barcode The internal barcode identifier for the row, e.g. internal01. external_barcode The external barcode identifier for the row, e.g. external01. Column titles are defined within the first row of the sample sheet. These must be defined in lowercase using the column title values listed above.
Sample sheet validation occurs against the hardware and between rows in the sample sheet to ensure validity when the sample sheet is loaded. Validation and importing the sample sheet requires all the flow cells to be used in the experiment. Be careful to ensure the correct flow cells and positions are used.
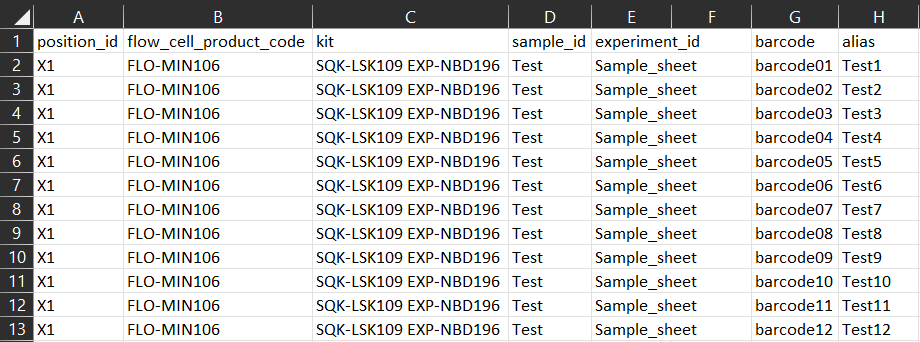
Below is an example sample sheet CSV file for using the Native Barcoding Expansion 96 (EXP-NBD196) with the Ligation Sequencing Kit (SQK-LSK109) on a GridION device for a single position.
-
Sequencing parameters using a JSON file
Alongside a CSV file containing the sample information, users can also upload a JSON file containing the sequencing parameters for their experiments rather than inputting the information manually.
The JSON file is exported from MinKNOW and contains all default sequencing parameters that can be altered:
Run options tab:
"runLengthHours": 72(Run length in hours condition. This must be an integer.)
"runLengthCondition": available_pores(Run length based on flow cell end of life condition. Run length in hours will be ignored if this condition is set.)
"targetDataCondition": estimated_basesorpassed_basecalled_bases(Run length based on target data output. This can be data estimated or data basecalled, respectively.)
"enrichDepleteAdaptiveSamplingEnabled": false,
"enrichDepleteAdaptiveSamplingRefFile": null,
"adaptiveSamplingChannelStart": 1,
"adaptiveSamplingChannelEnd": 512,
"enrichDepleteAdaptiveSamplingBedFile": null,
"shouldEnrichAdaptiveSamplingRef": true,
"barcodeBalancingEnabled": false,
"barcodeBalancingCustomBarcodes": false,
"barcodeBalancingBarcodeSelection": null,
"minReadLength": 200,(Read length selection. This can be 20, 200 or 1000 bp)
"activeChannelSelection": true,
"muxScanPeriod": 1.5,
"groupChangePeriod": 16,
"reservedPores": true,Analysis tab:
"basecallingEnabled": true,(Basecalling can be switched on/off as true/false)
"modifiedBasecallingEnabled": false,(Modified basecalling can be switched on/off as true/false. To enable methylation calling, this needs to be enabled.)
"barcodingEnabled": true,(Barcoding can be switched on/off as true/false)
"basecallModel": "dna_r9.4.1_450bps_hac.cfg",(Basecalling model depends on the flow cell and kit combinations. To enable methylation calling, this needs to be enabled.)
"modifiedBasecallingContext": "",(To enable methylation calling, this needs to be enabled)
"trimBarcodesEnabled": true,(Trim barcodes can be switched on/off as true/false)
"requireBarcodesBothEnds": false,(Requirement to have barcodes on both ends can be switched on/off as true/false)
"detectMidStrandBarcodes": false,(Detect mid read barcodes on/off as true/false)
"overrideMidBarcodingScore": false,(Override mid barcoding score can be switched on/off as true/false)
"overrideBarcodingScore": false,(Override barcode score can be switched on/off as true/false)
"minBarcodingScore": 60,(Selection of barcode score. This can be an integer between 40 to 100)
"minBarcodingScoreMid": 50,(Selection of mid barcode score. This can be an integer between 40 to 100)
"alignmentRefFile": null,
"alignmentBedFile": null,Output tab:
"dataOutputLocationType": 0,
"offloadOrOutputLocationNavParams":
"path": "/data/."
"fastQEnabled": true,(FASTQ files can be switched on/off as true/false)
"fastQReadsPerFile": 4000,(Number of reads per file. This is an integer)
"fastQDataCompression": true,(FASTQ compression can be switched on/off as true/false)
"fast5Enabled": true,(FAST5 files can be switched on/off as true/false)
"fast5ReadsPerFile": 4000,(Number of reads per file. This is an integer)
"fast5DataCompression": "vbz_compress",(Compression type. This can be"vbz_compress"or"zlib_compress")
"selectedRawOutput": "fast5",
"pod5Enabled": false,
"pod5ReadsPerFile": 4000,
"bamEnabled": false,(.BAM output can be switched on/off as true/false. To enable methylation calling, this needs to be enabled.)
"bamWriteMultiple": true,(read length in kb)
"readFilteringEnabled": true,
"readFilteringMinQscore": 9,
"readFilteringMinReadlength": null,
"readFilteringMaxReadlength": null,(read length in kb)
"readSplittingEnabled": false,
"overrideMinReadSplittingScore": false,
"minReadSplittingScore": 58,
"bulkFileEnabled": false,
"bulkFileRaw": "1-512",
"bulkFileEvents": "1-512",
"bulkFileReadTable": "1-512",
"bulkFileRawEnabled": false,
"bulkFileEventsEnabled": true,
"bulkFileReadTableEnabled": true -
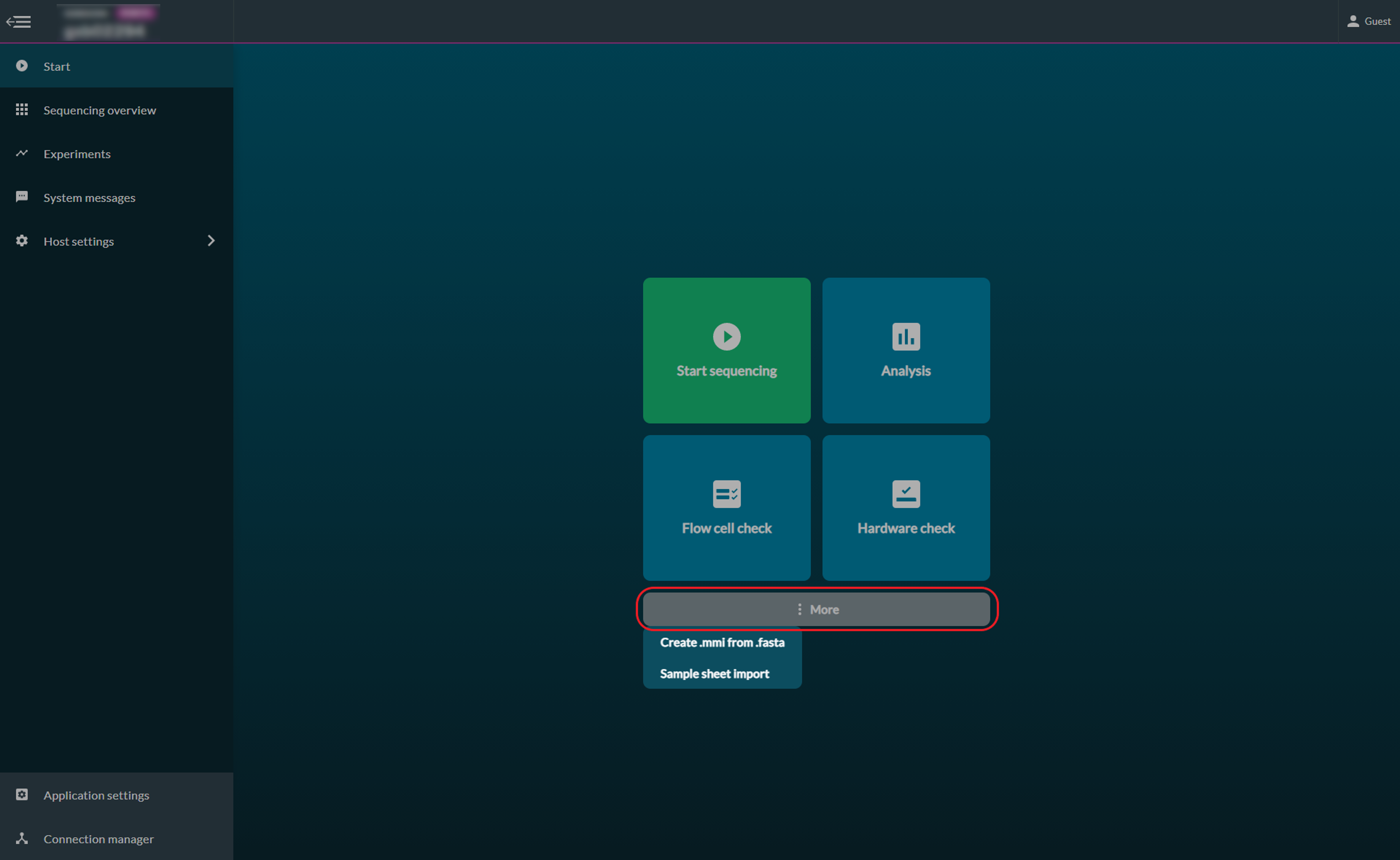
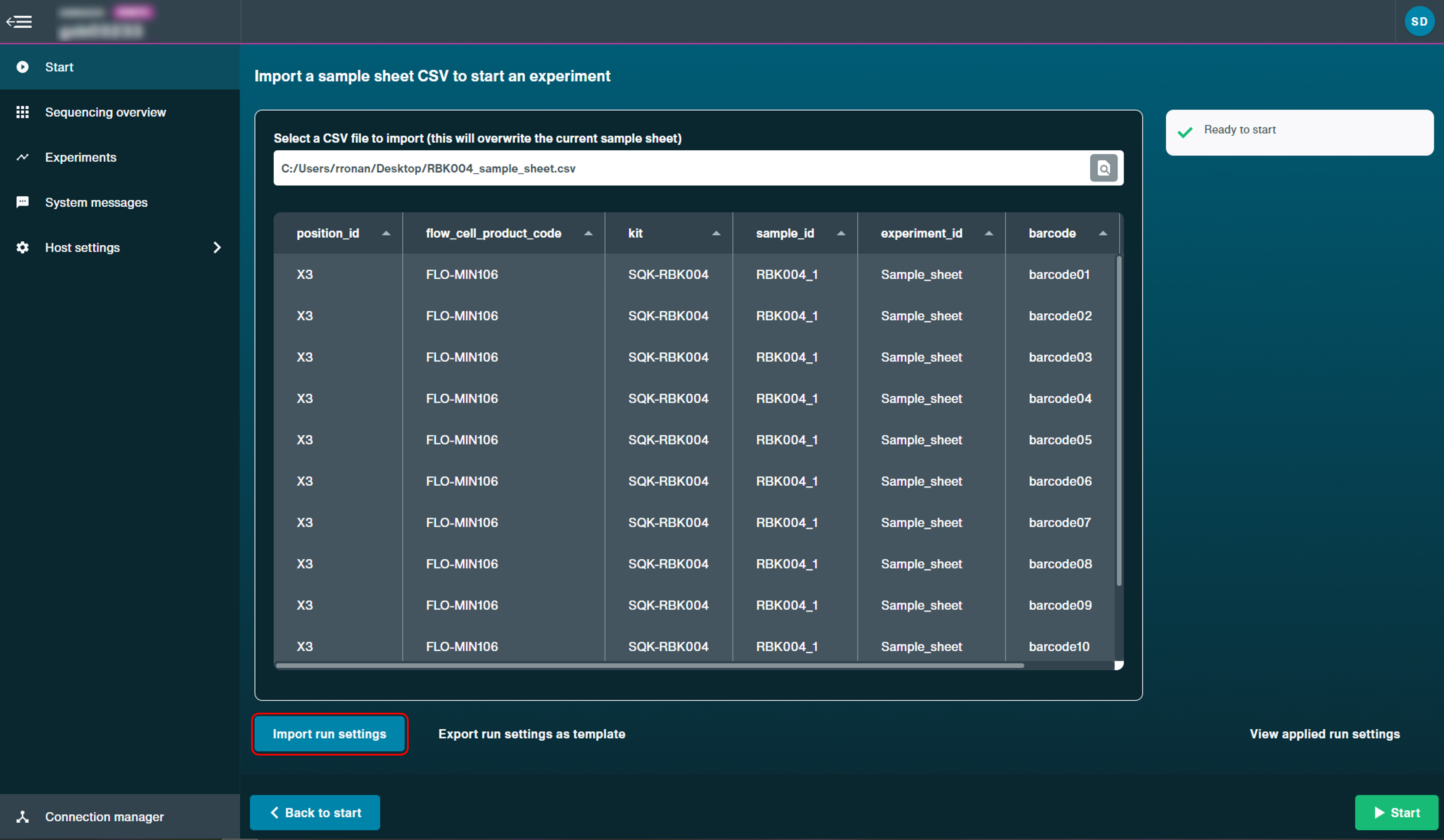
Click 'More' and click 'Sample sheet import'.
-
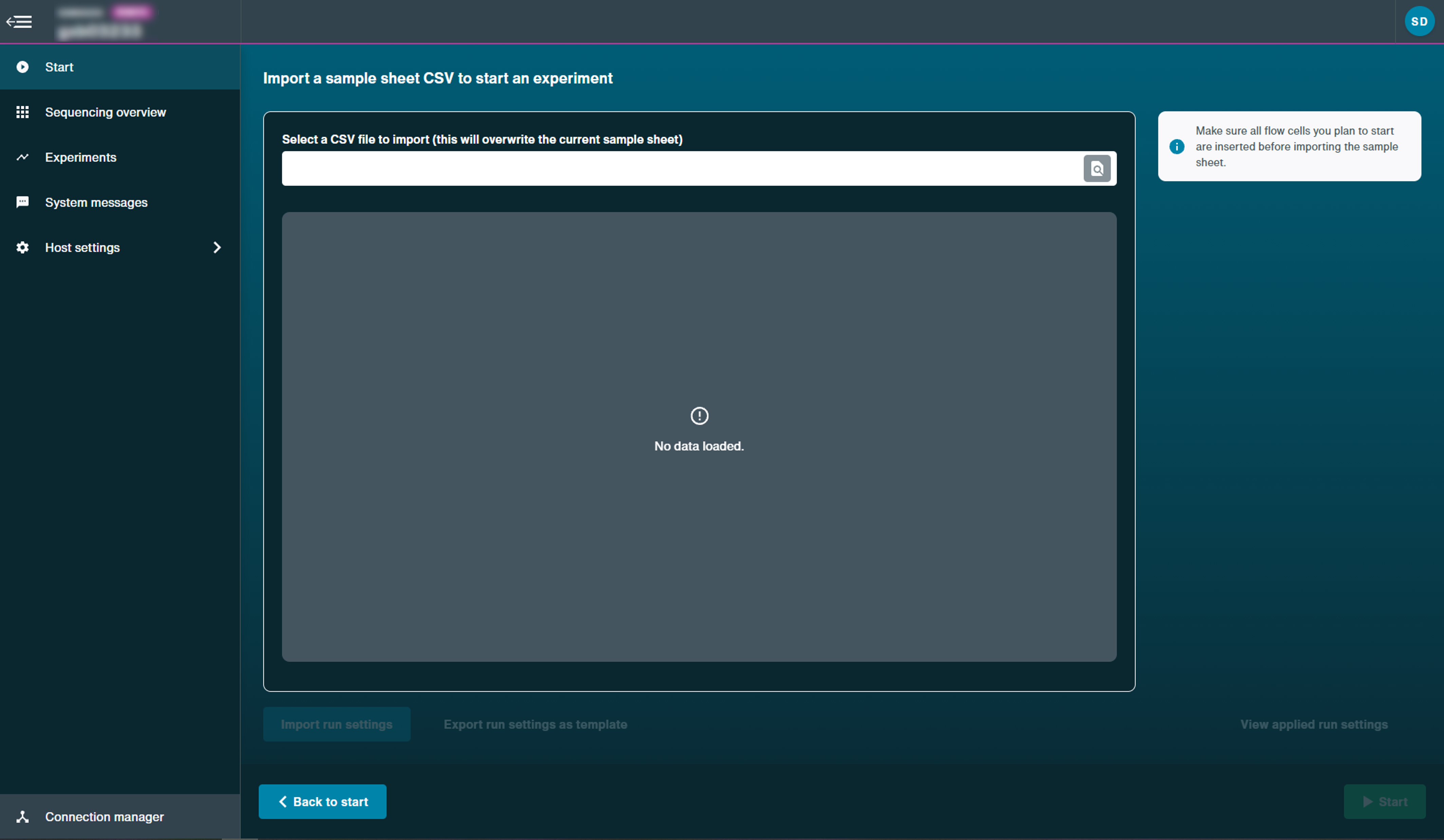
Select and upload the CSV file.
-
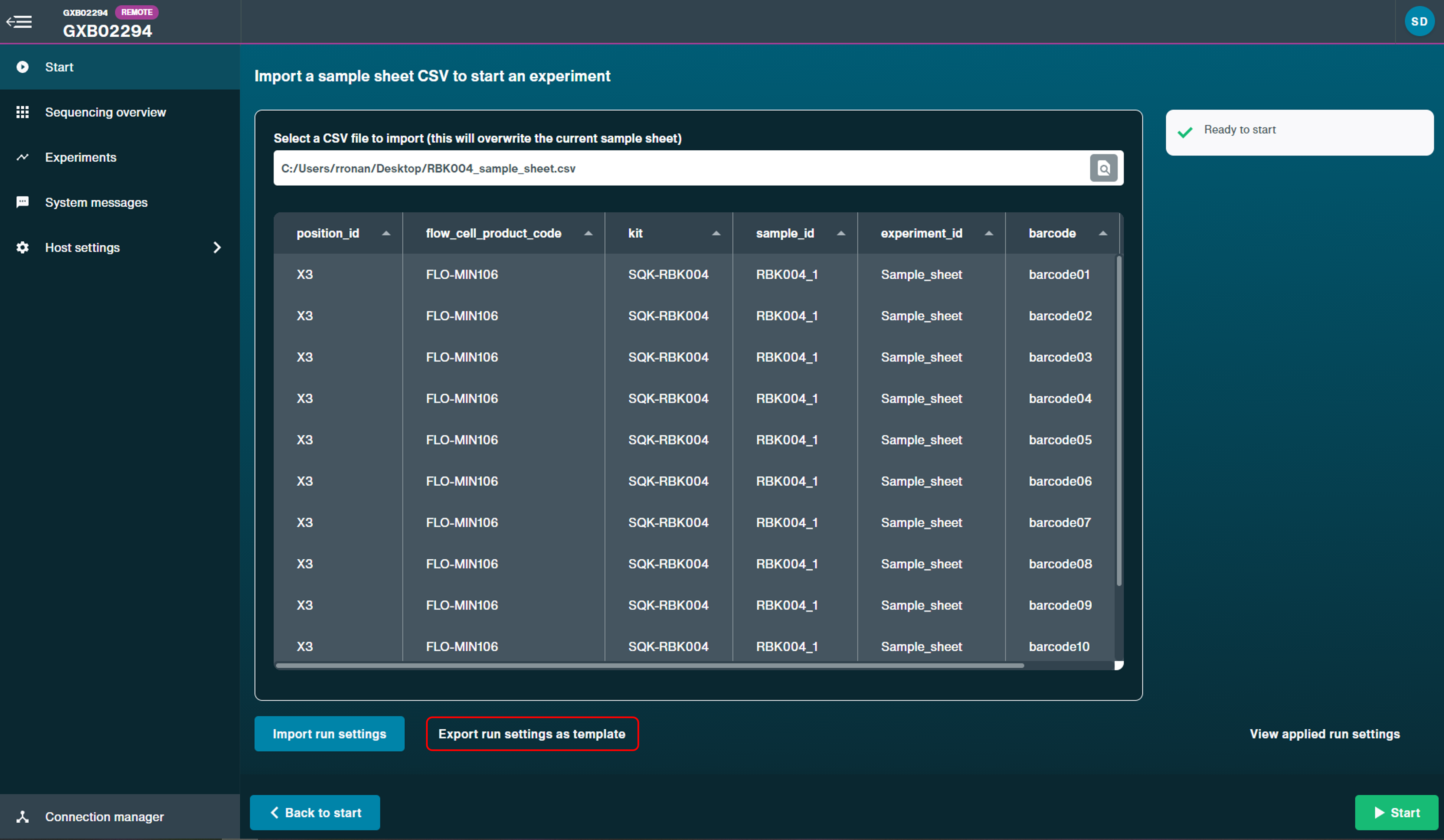
Click 'Export run settings as template' when the CSV file has been uploaded.
-
Open the exported JSON file to edit sequencing parameters, as described above and save.
-
Upload the JSON file by clicking 'Import run settings'.
-
Click 'Start' to start the run using the CSV and JSON files.
-
Error messages that you may see if you make a mistake in the sample sheet:
- Position defined using
position_iddoes not exist - Position defined using
flow_cell_idcannot be found flow_cell_idandposition_idare used but theflow_cell_iddoes not match that on the EEPROM (if there is a value on the EEPROM)flow_cell_product_codeis used but the value doesn't match the EEPROM value (if there is a value on the EEPROM)- Different
sample_idvalues are assigned against the same position whilst setting up a run flow_cell_product_codeandkitdo not match- Zero or more than one sequencing
kitlisted in kit - Barcode kits in
kitdo not exist as an option on the selected protocol typemust be a valid selection from the list of valid types outlined aboveexperiment_idmust exist within each row and must all have the same value
- Position defined using




