-
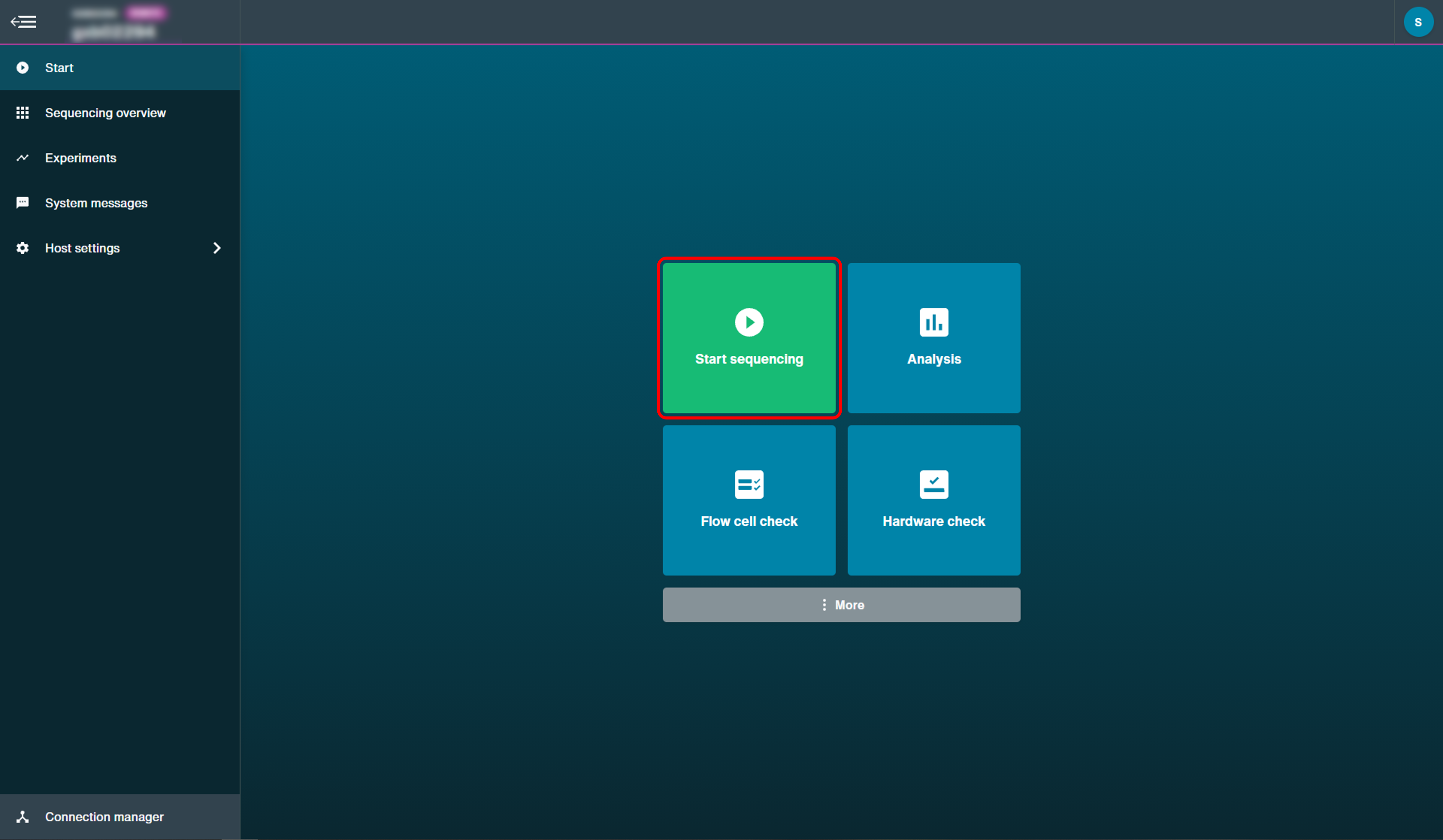
Click 'Start Sequencing' on the Start page to set up the sequencing parameters for your experiment.
-
Optional actionWhen running multiple flow cells simultaneously, it may be preferred to upload sample names and corresponding flow cell positions from a CSV file, rather than manually.
Please see Sample sheet upload for instructions.
-
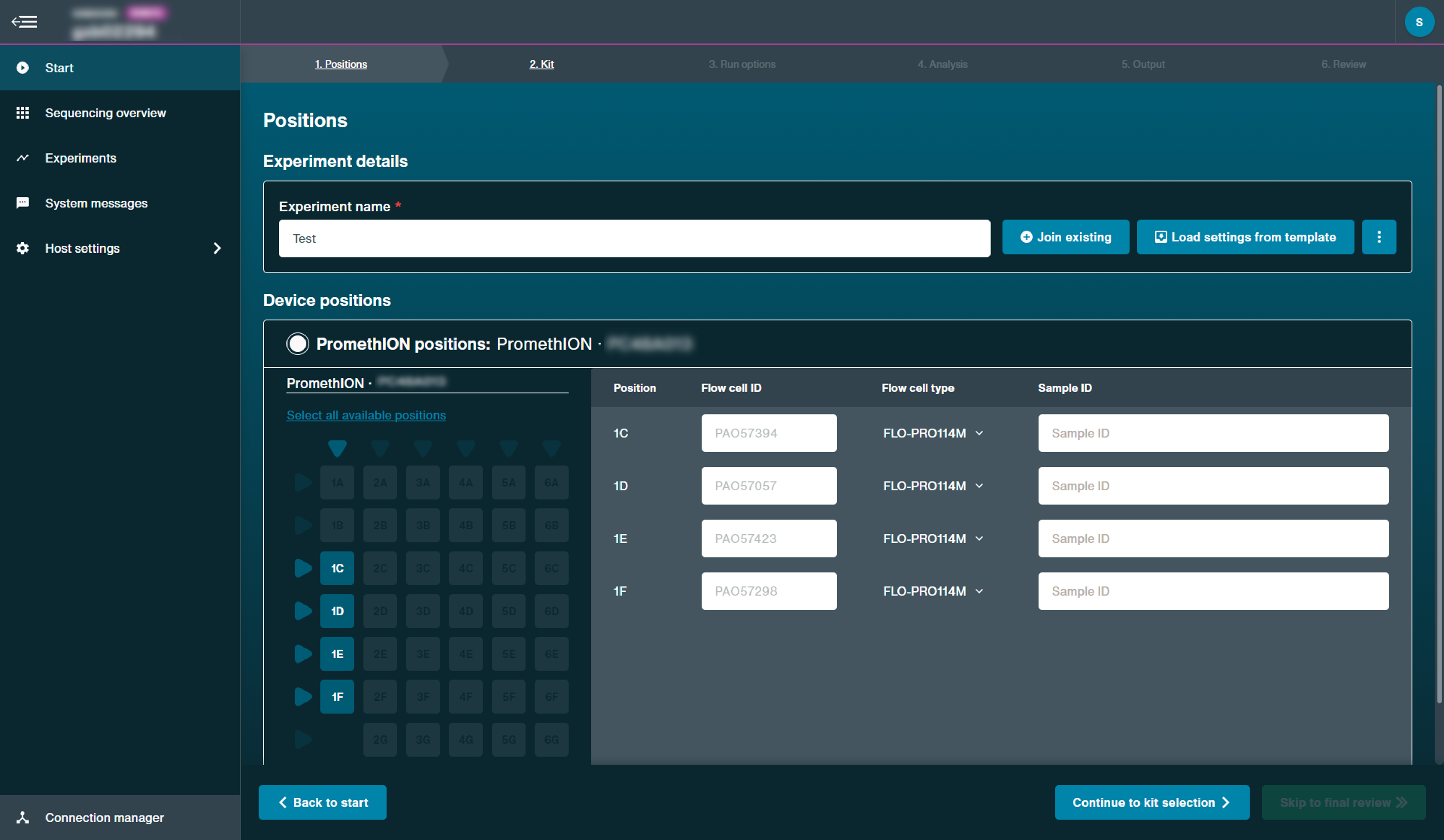
Type in the experiment name, sample ID and choose flow cell type from the drop down menu.
Select Continue to Kit Selection to move to the next page.
Ensure the experiment name and sample ID does not contain any personally identifiable information.
To use previously saved settings, please see the "Save sequencing run settings" page of this protocol.
Use "Select all available" to select all the connected flow cells or use the diagram above to select specific flow cells for an experiment.
Note: If sample ID is not filled in, there will be no sample ID in the folder structure.
-
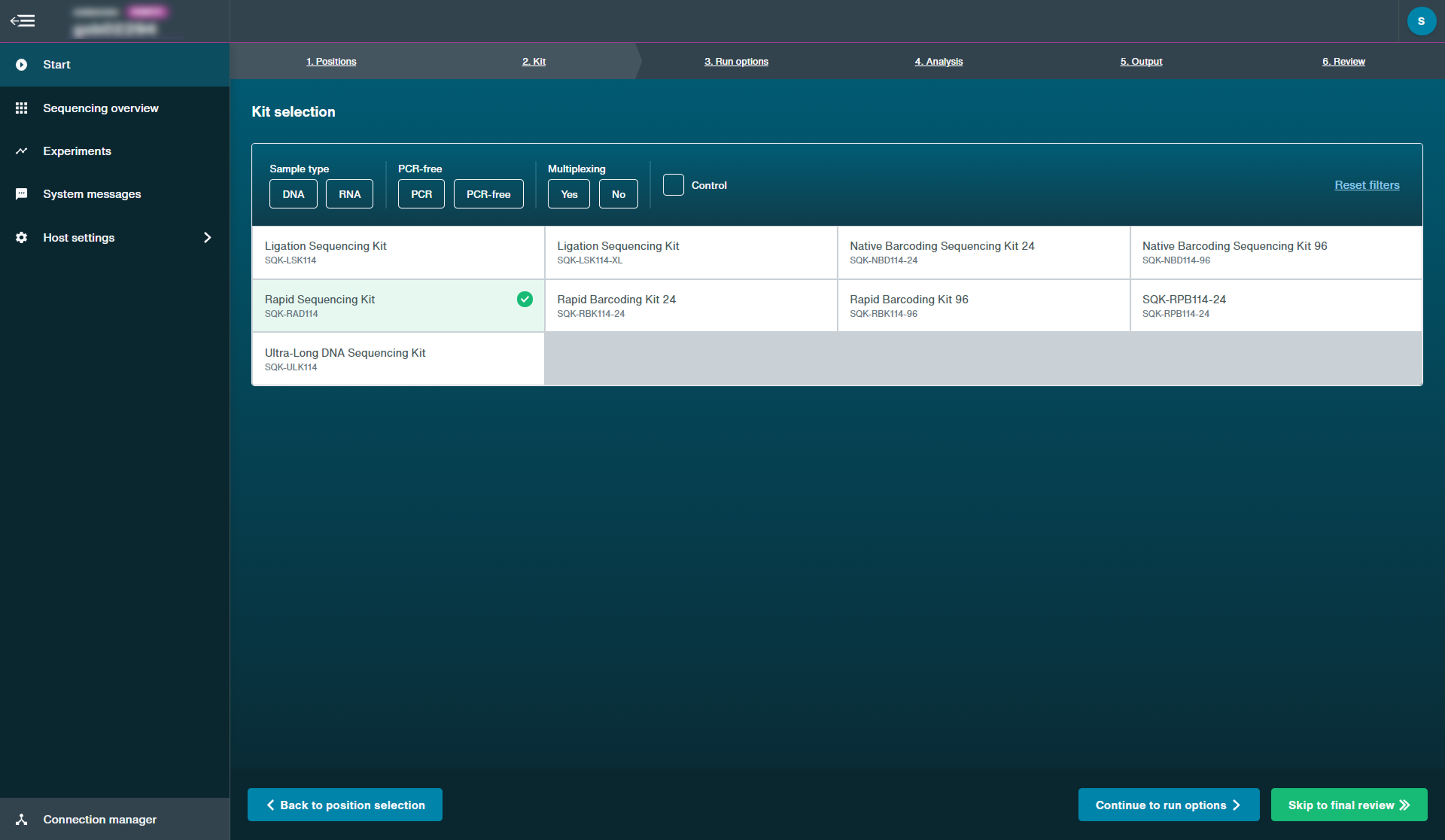
Select the kit and expansion(s) used to prepare the library.
One sequencing kit can be selected as well as one or more expansions. If a sequencing kit does not have any associated expansions, no expansion options will appear.
The filter options may be used to find the kit used. For example, click DNA to filter for DNA sequencing kits.
For control runs, there is a Control checkbox which only filters for the control experiments of the kits. This will automatically set the sequencing parameters for a control run.
Note: Run length will be reduced to 6 hours for control experiments.Click Continue to Run Options to choose run parameters.
-
To skip setting up the other parameters, click Skip to final review.
You can also return to any previous pages by using the Back to... button.
We recommend keeping the default sequencing parameters unless you have a specific need to change them.

-
Set up the run options of the sequencing run.
The run options tab provides variables for run time, minimum read length and the option to use adaptive sampling.
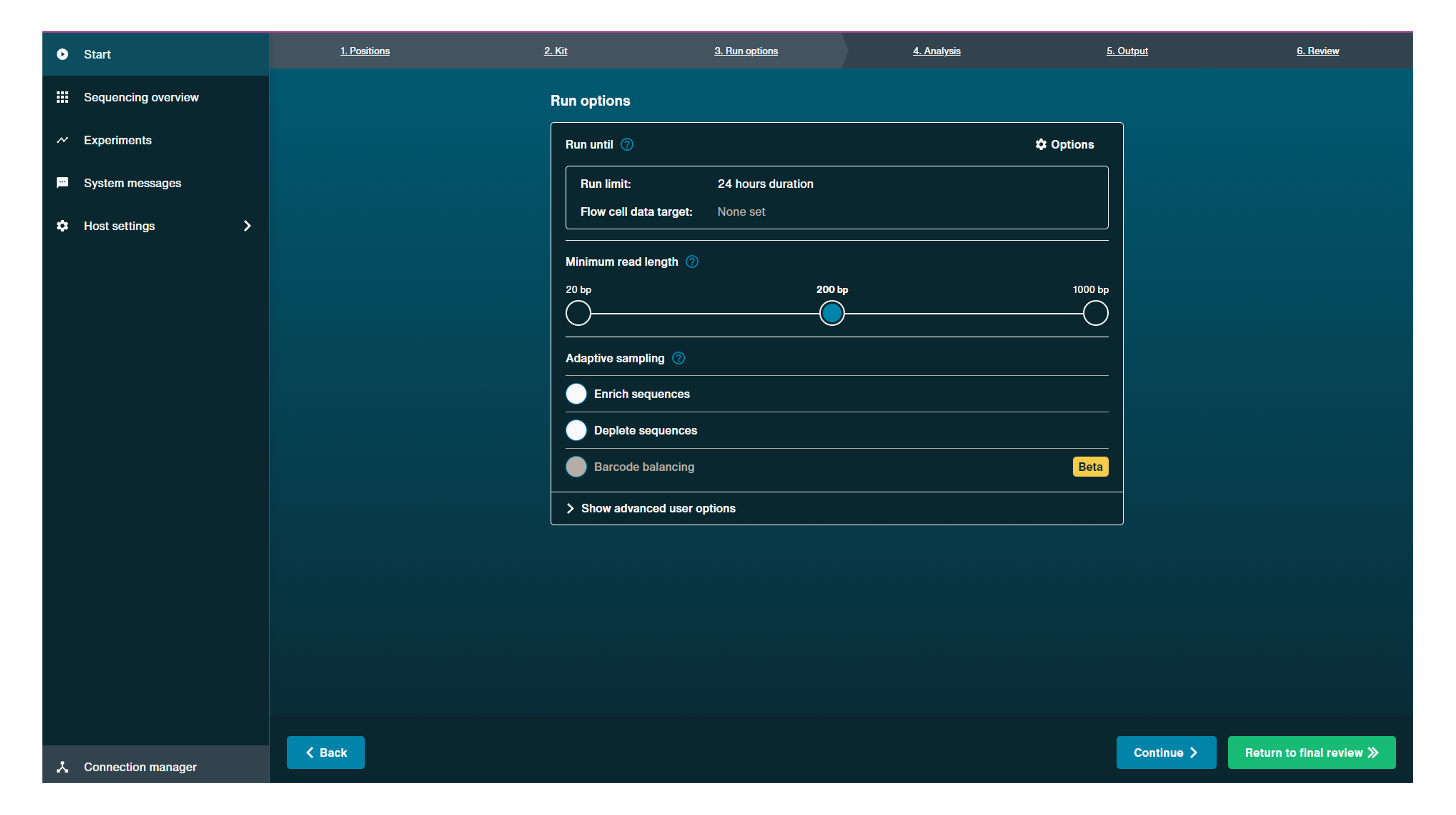
Run until:
Set up the rules to define when a sequencing run will stop. If multiple rules are set up, the first rule met will stop the run.
Click Options to open run until options.
Run limit: This rule must be defined based on run time length in hours or when a flow cell is end of life and has no more sequencing pores available.
Use the drop-down menu to choose:
- "Time equals" and hours
- or "Flow cell is" which will automatically be set as "End of life".
Flow cell data target: This rule can be used to define a target amount of data to reach before a sequencing run is stopped. This can be specified for either estimated or basecalled bases and your data target will be available to view on the experiment graphs to allow you to monitor data progress.
Please note, basecalling must keep up with data output in real-time. If the device or basecall model will not keep up and reach the target output before the run limit rule is met, we recommend using the "Estimated bases" option. However, if your experiment will not reach the target data before the end of the experiment as defined by the run limit rule, you will be warned by the UI.
Use drop-down menu to select for "Estimated bases equal" or "Basecalled bases equal" and input the value in either Gb or Mb.
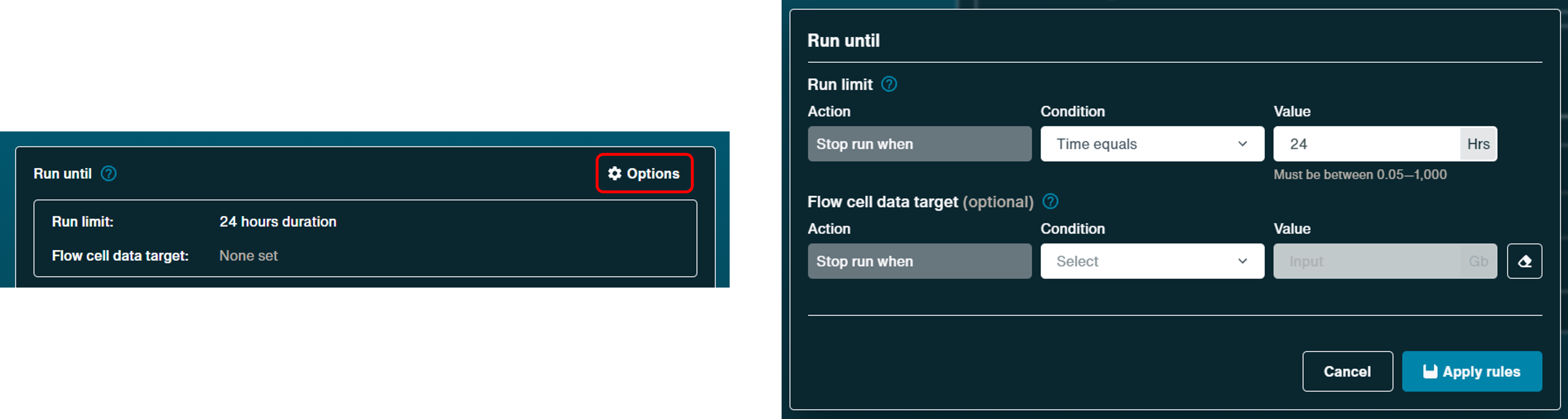
Minimum read length:
Define the preferred minimum read length, from as short as 20 bp, up to 1000 bp using the sliding scale. This directs the software to write sequencing files from the minimum size read length selected.
Please note, the lower the minimum read length, the more data is produced due to a wider range of fragment lengths which can be processed.
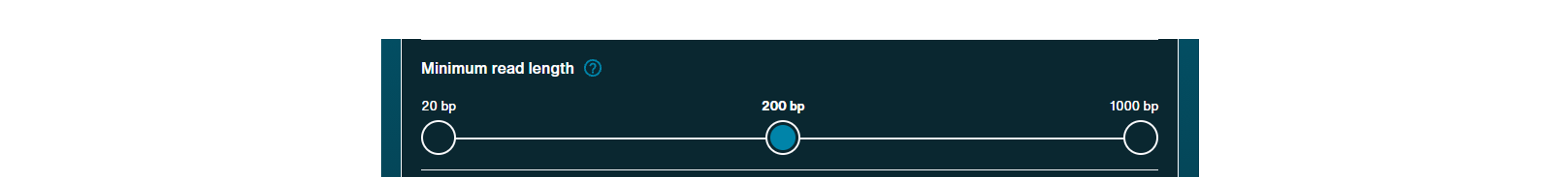
Adaptive sampling:
Choose to select for or reject specific sequences in real-time by selecting "Enrich or deplete sequences". Upload an alignment reference and any specific sequence coordinates of interest and define whether to enrich or deplete alignment matches.
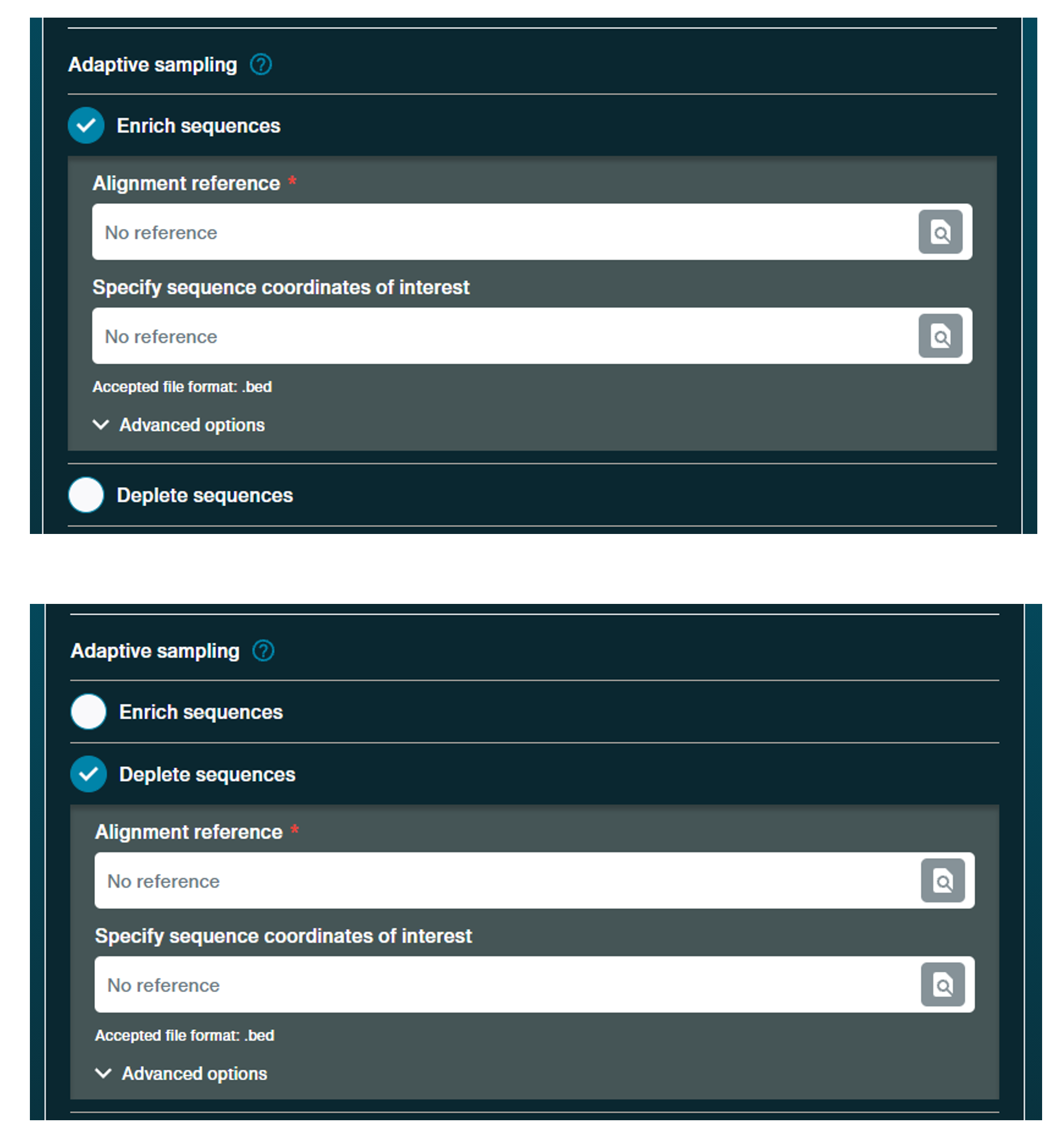
There is also beta support for barcode balancing. Under this option, there will be options to balance all barcodes detected or to choose barcodes to balance.
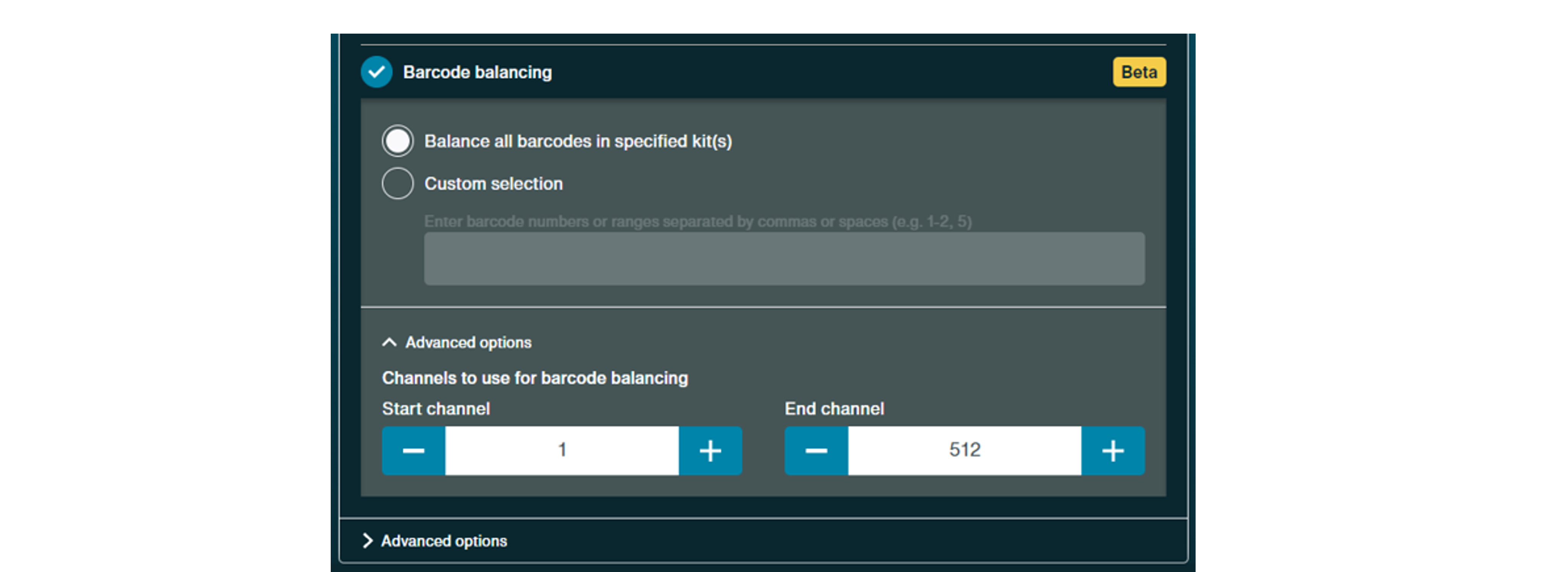
For more information on adaptive sampling, please see our info sheet:
- Adaptive Sampling info sheet
Please note, MinION Mk1C does not support running live basecalling alongside adaptive sampling. Please see basecalling caveats in the Adaptive sampling info sheet in the 'Design and running an experiment with adaptive sampling' section.
Note: We recommend using alignment on bacterial-sized genomes.
Select Continue to analysis to proceed to the next section.
-
Active channel selection:
Active channel selection refers to a feature where if a pore is in the 'Saturated' or 'Multiple' state, the software instantly switches to a new pore in the group. If a pore is 'Recovering', MinKNOW will attempt to revert the pore back to 'Pore' or 'Sequencing' for ~5 minutes, after which it will select a new pore in the group. This maximises the number of pores sequencing at the start of the experiment.By default, this option is on but can be switched off. Time between pore scans can also be altered.
Reserved pore:
The reserve pore feature prioritises consistency and accuracy over immediacy by reserving wells where voltages have dropped until later in the run, such that other wells can catch up. Switch off this feature to fully front-load sequence data acquisition.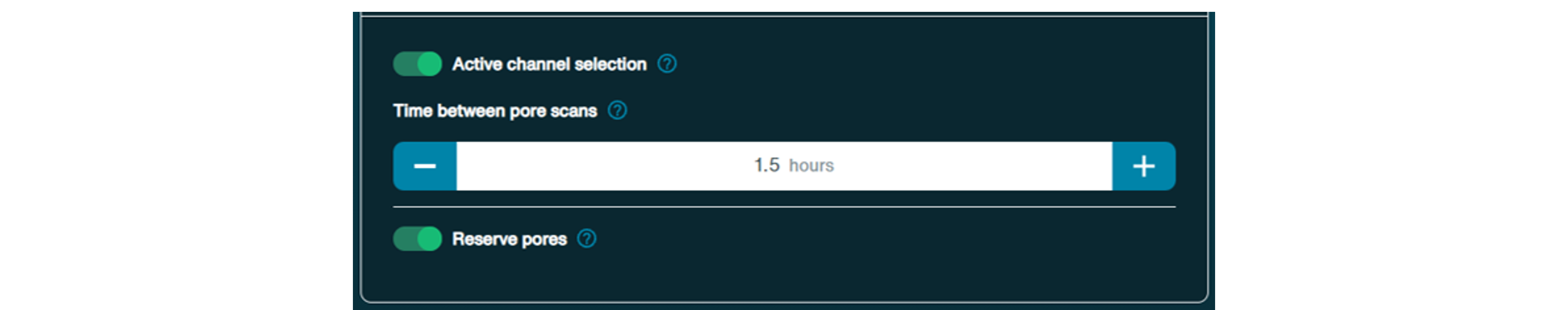
-
Choose basecalling, barcoding and alignment options.
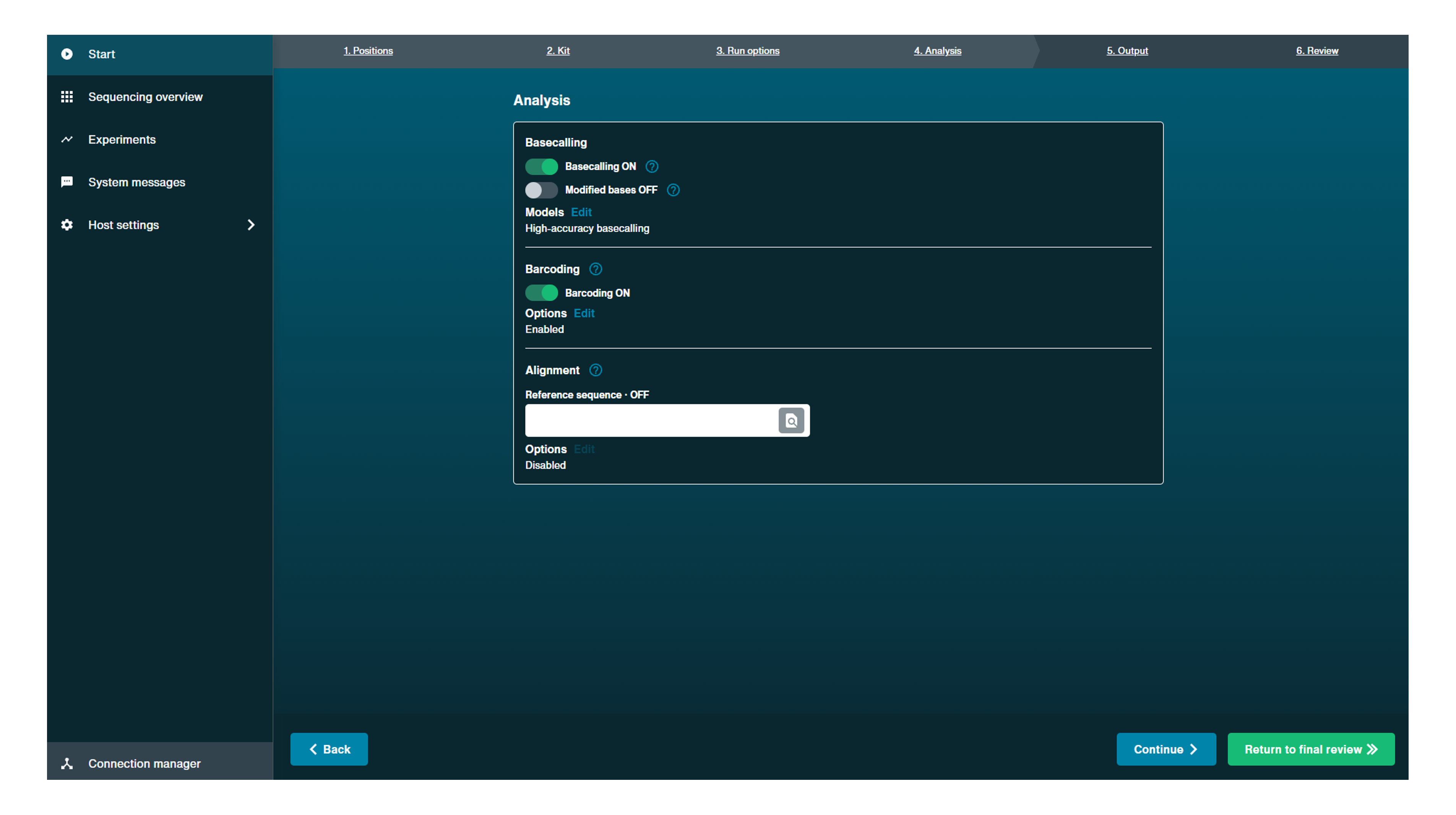
Basecalling:
This is on by default to perform real-time basecalling. This can be switched off and basecalling performed post-sequencing.
Note: If basecalling is disabled, barcoding and alignment will not be performed during sequencing.
Modified bases is off by default. This can be switched on to use the CpG context models and basecall 5mC and 5hmC.
To specify your basecall model and modified bases, click Edit options to open basecalling options dialogue box.

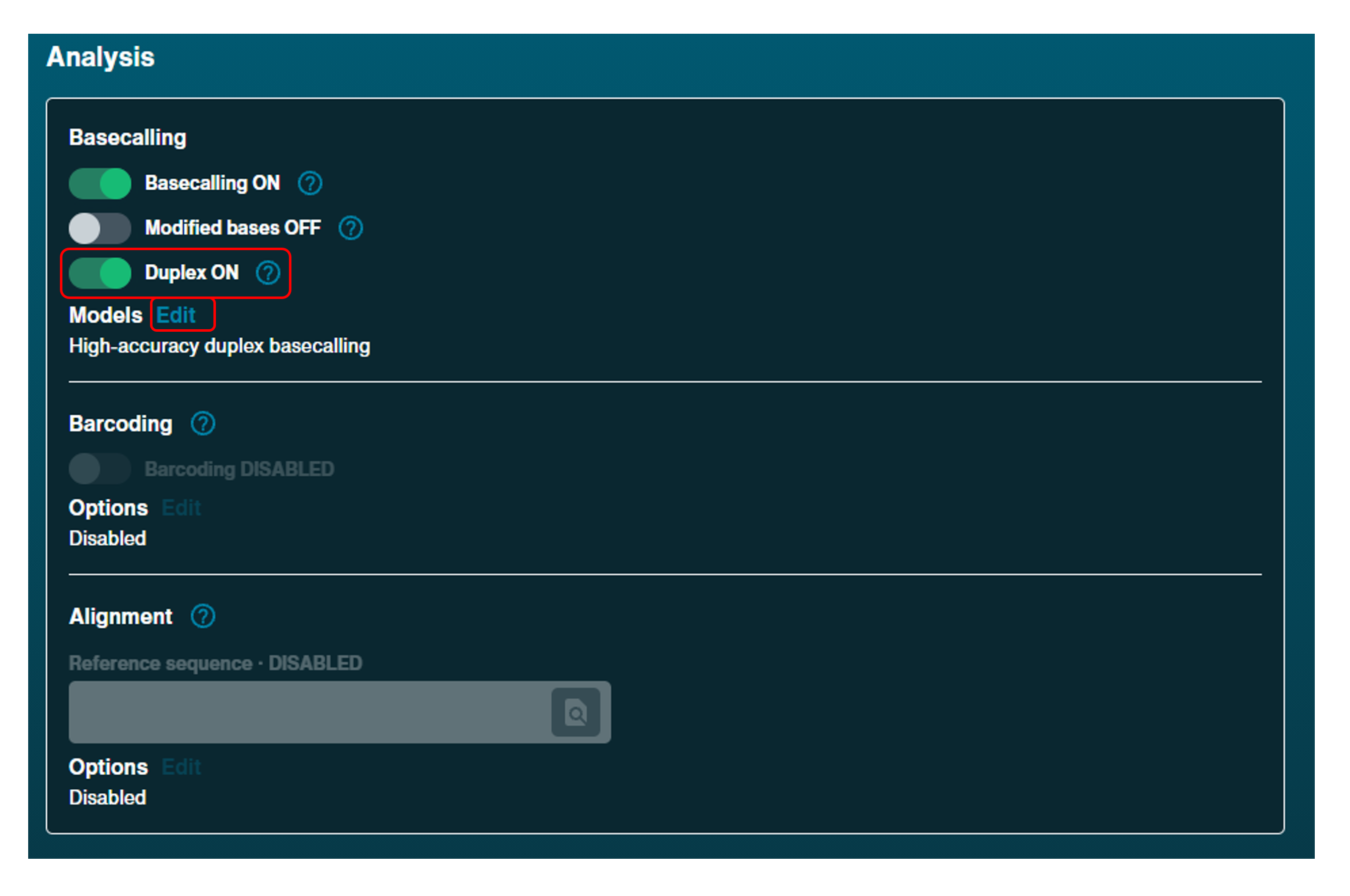
Duplex basecalling:
For duplex sequencing, toggle on duplex basecalling and click Edit options and select Duplex (HAC) in the dialogue box.Note: Duplex is only compabitible with R10.4.1HD flow cells and the SQK-LSK114 kit. Barcoding, alignment, modified basecalling, adaptive sampling and run until are not currently available.
Barcoding:
Barcoding options are only available when a barcoding sequencing kit or expansion has been selected. Barcoding can be switched off and performed post-sequencing.
To specify the barcoding options, click Edit options to open the barcoding options dialogue box. From here, barcode trimming, mid-read barcoding filtering and barcoding minimum score can be defined.
By default, barcoding trimming is off. This can be enabled to remove barcodes from demultiplexed reads. Please note, some primer sequences may also be trimmed together with the barcodes.
Minimum barcoding score can be increased to improve confidence in barcode detection classification at the expense of more unclassified reads.
Each barcode demultiplexed by the basecaller will have its own folder and can be processed separately:
/data/fastq_pass/barcode[01-XX]
/data/fastq-fail/barcode[01-XX]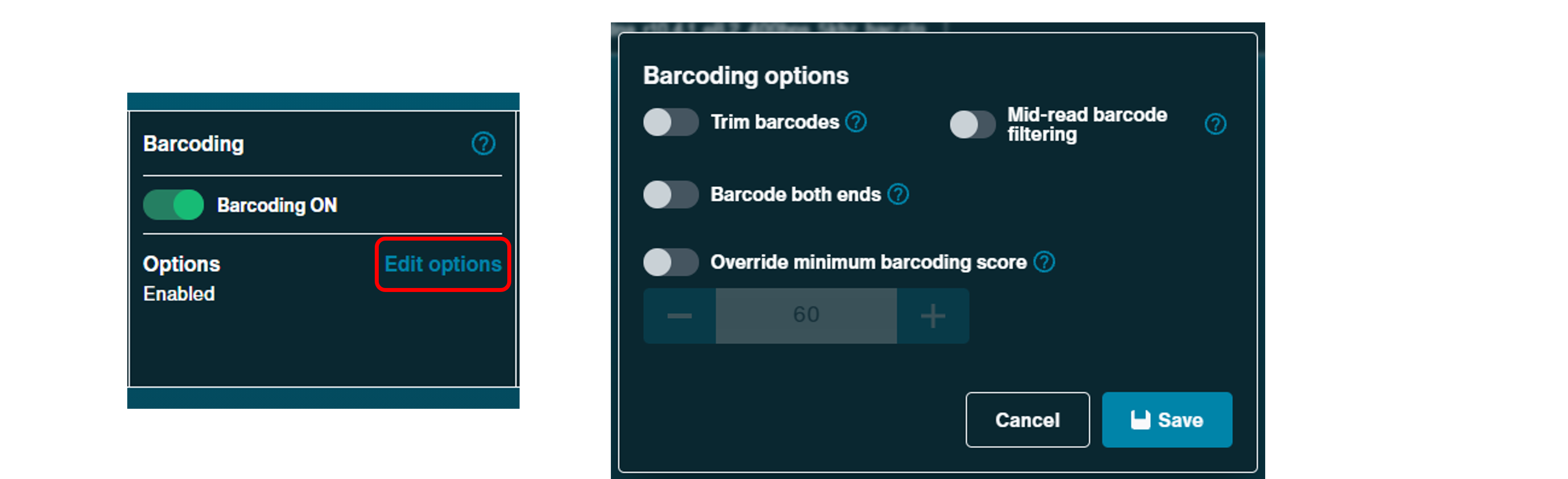
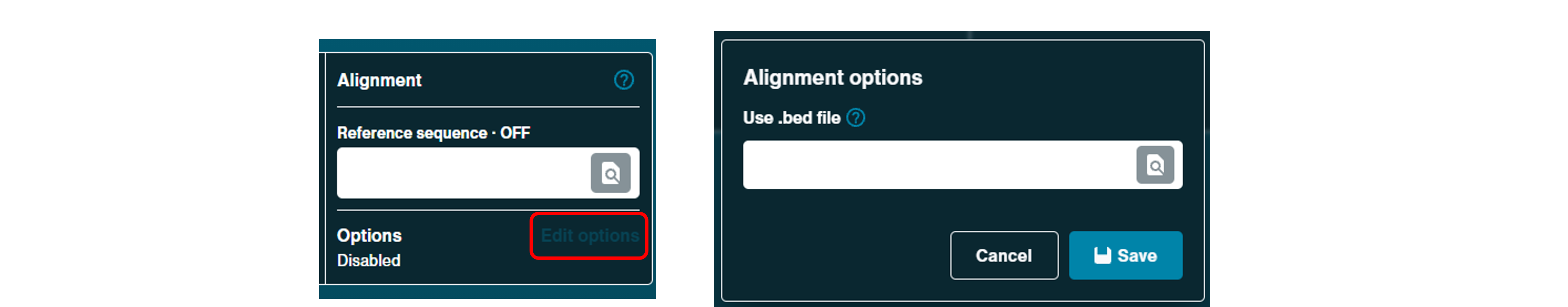
Alignment:
To use alignment during sequencing, upload an alignment reference file as a .fasta or .mmi file. We recommend uploading a reference locally on bacterial-sized genomes. Uploading a file can take a few minutes and is compute-dependent.
A reference file can contain multiple entries in the same file (e.g. multiple chromosomes) and alignment hits from these files are used to populate the alignment graphs which can be viewed on the GUI.
A BED file can also be uploaded alongside the reference when there is a specific interest in a particular region of the reference (e.g. specific gene in a chromosome). Alignment hits from BED files will be highlighted in the sequencing .txt file generated in the data folder. Click Edit options to open a dialogue box to upload a BED file.
Note: For barcoding or alignment to be performed during sequencing, basecalling must be enabled.
Select Continue to Output to proceed.
-
Basecall models
Basecalling models can be selected at two stages in the MinKNOW software:
- Real-time basecalling: Basecalling model can be selected when setting up a sequencing run on the "Run options" page of MinKNOW. This is basecall the sequencing run in real-time.
- Post-run basecalling: Basecalling can be set up post-sequencing run on MinKNOW. Instructions can be found in the "Post-run basecalling" section of this protocol.
Dorado is the basecaller server in MinKNOW which provides multiple models for basecalling nanopore data.
There are four options for model selection in the drop down menu:
1. Fast basecalling - This model is able to keep up with a high-throughput sequencing experiment on a MinION Mk1B, MinION Mk1C, GridION or PromethION.
2. High-accuracy basecalling (HAC) - This provides a higher single-molecule accuracy than the Fast model. It is currently 5-8 times more computationally-intensive than the Fast model, so users should ensure their data transfer and device utilisation is scaled appropriately for this.
3. Super-accurate basecalling (SUP) - The Super accurate model has an even higher single-molecule accuracy, and is ~3 times more intensive than the HAC model.
4. Super-accurate basecalling - Plant - The super accurate model for plant basecalling, trained using Maise on R9.4.1 E8.1.
5. Duplex basecalling - This enables basecalling of the complement and template strand directly on MinKNOW without using command line. Both high-accuracy and super-accurate basecalling is available with duplex when using a High Duplex Flow Cell (R10.4.1HD).
6. No basecalling
Note: GPU devices will basecall in near real-time; CPU devices, like a standard laptop for MinION Mk1B, will not maintain real-time basecalling.
-
Select the output data location, format and filtering options.
The output tab provides variables for data output including file name, location and format.
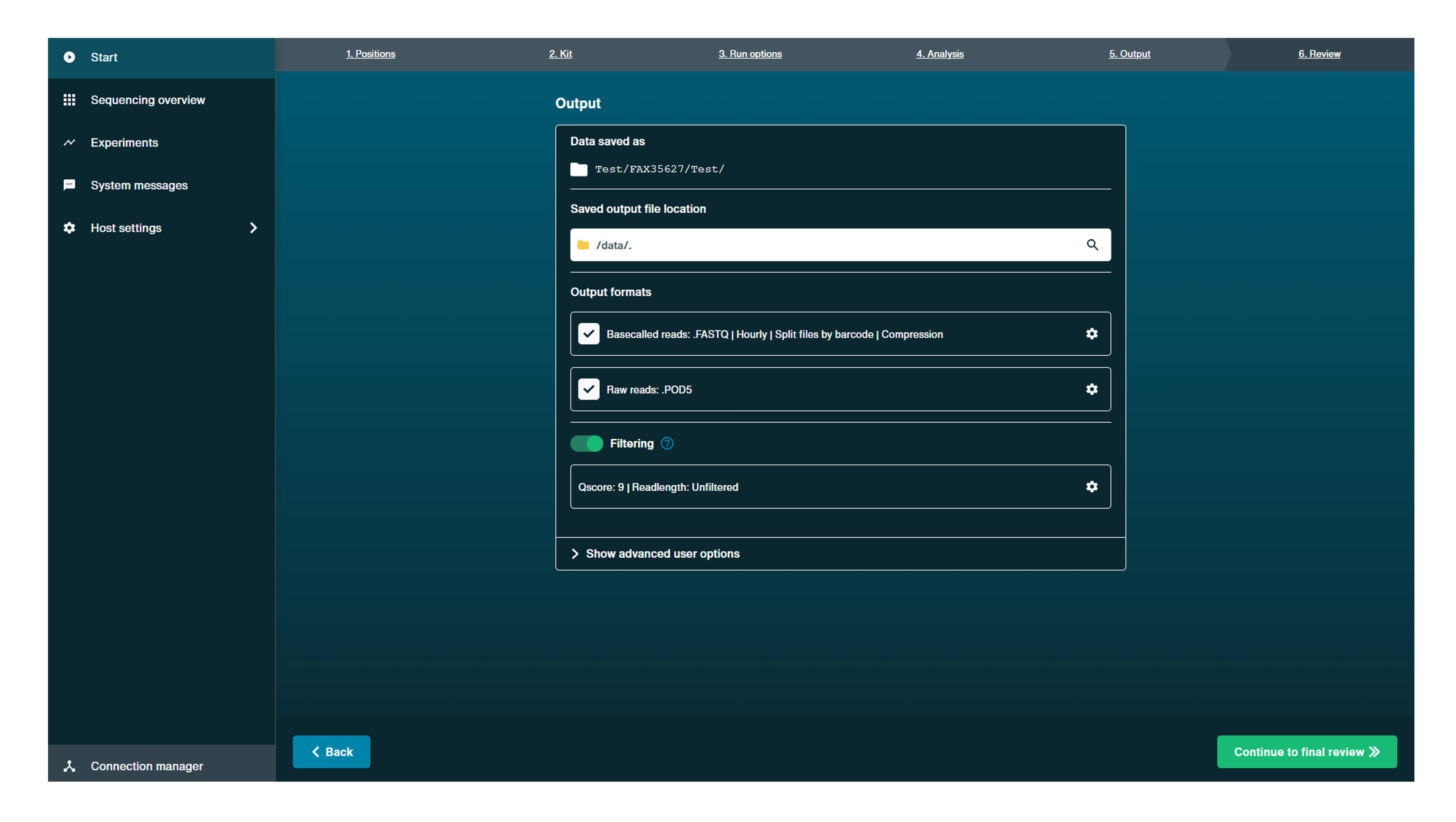
Data saved as:
Check the name of the output file for the data.
Output location:
Confirm the location for the output data file. Click the file path name to open a pop-up to alter file path or create a new file.
Click Choose location on the pop-up to confirm new file location.
Output location storage capacity:
A bar indicator is displayed below the selected data output file location. This bar will display red when the disk space used reaches more than 80% of the total available disk space. Otherwise, the bar will display green. Below the bar, you can see how much drive space is available.
Output format:
Confirm output file type and file compression using the checkboxes.
Note: Below, reads per file can be changed for both raw reads and basecalled reads, but take note:
- Fewer reads per file: reads become more quickly available. However, too few reads per file means MinKNOW may not keep up with writing out files in real-time.
- More reads per file: reduces the number of files especially for amplicon/cDNA experiments that produce a large number of reads. Some downstream analysis tools may have an upper limit on the uploaded file size.
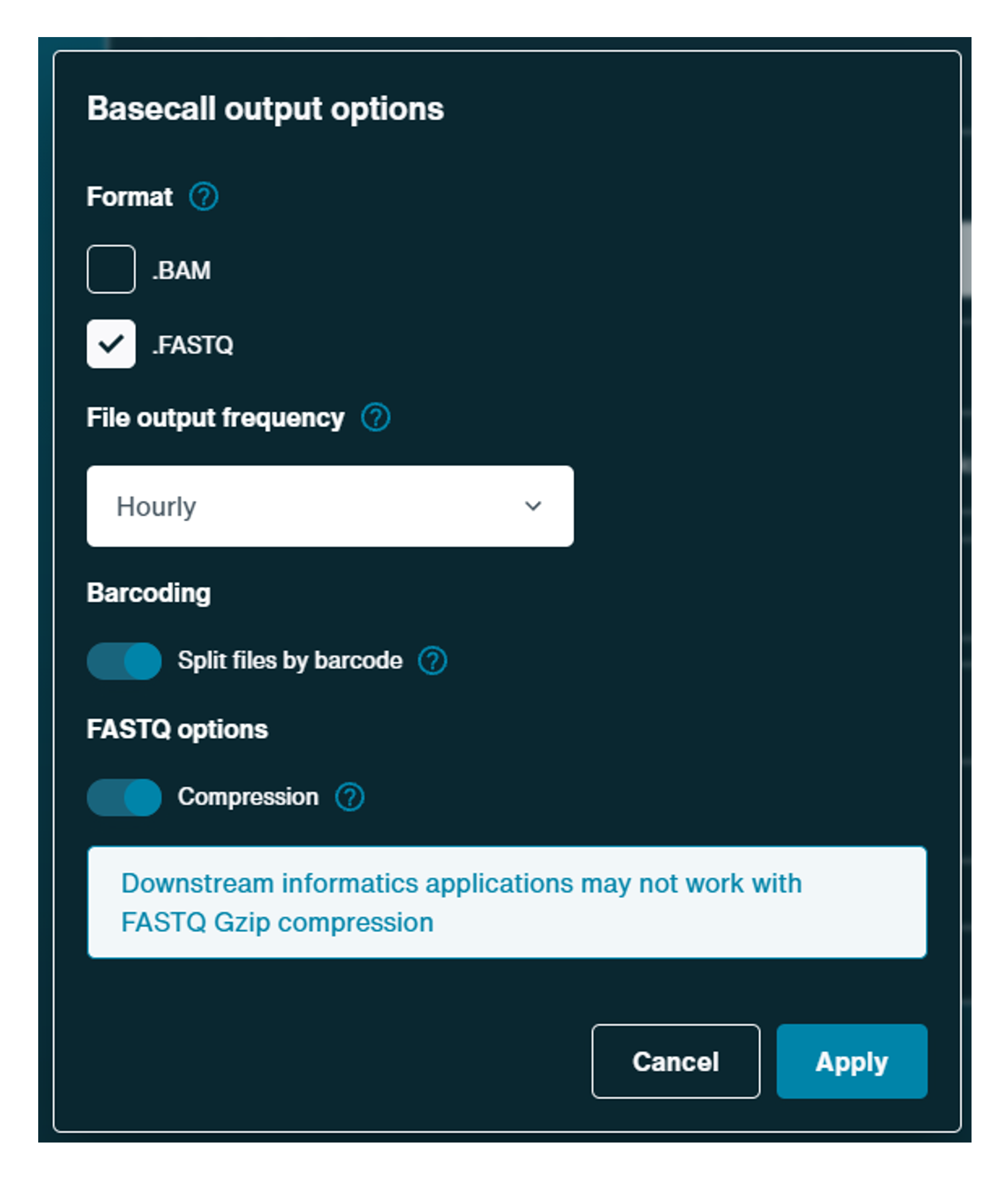
Basecalled reads:
The .FASTQ will be on as default if basecalling in real-time.Click the cog to open further settings:
- File output frequency can be adjusted for basecalled files. These can be produced every 10 minutes (many small files), hourly (fewer larger files) or at the end of the run (single large file).
- For barcoding runs, basecalled reads are split into files by barcode by default.
- Gzip compression of FASTQ files is on by default of basecalled reads. Note: FASTQ Gzip files may prevent some downstream informatics applications.
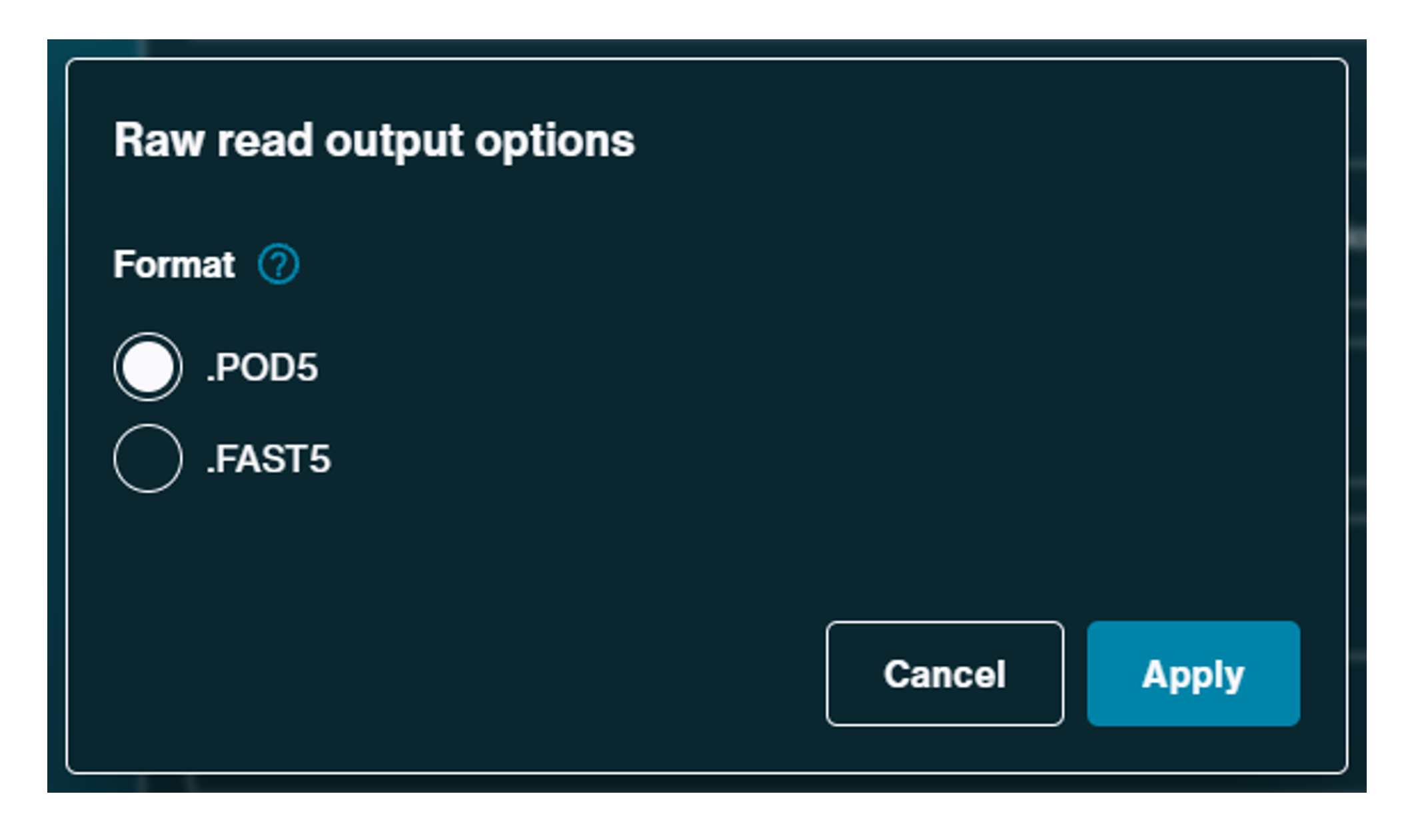
Raw reads:
Raw reads output can be saved as .POD5 or .FAST5.- For Kit 14 chemistry and SQK-RNA004, .POD5 is the default file output. The .POD5 output generates files hourly.
- The .FAST5 output file is the default format for our previous and "legacy" chemistry kits. The.FAST5 output produces files for every 4000 reads.
.POD5 is a Nanopore-developed file format which stores Nanopore data in a more accessible way and can be used as an alternative to FAST5 output. This output also writes data faster, uses less compute and has smaller raw data file size.
We recommend keeping the default settings. However, the raw read outup files can be changed by clicking the cog.
Aligned reads:
The .BAM output is on as default for sequencing runs using live alignment.
This can be observed in the settings by clicking the cog next to Basecalled reads.Filtering
Filtering options can be used to determine which reads are classed into pass or fail files. These options may also be used to determine which predefined reads, read lengths and Qscore during basecalling can be split out in some live graphs.
Click the cog to open the filtering options pop-up and use the GUI to alter Qscore and to define minimum and maximum readlength for a pass read, if required.

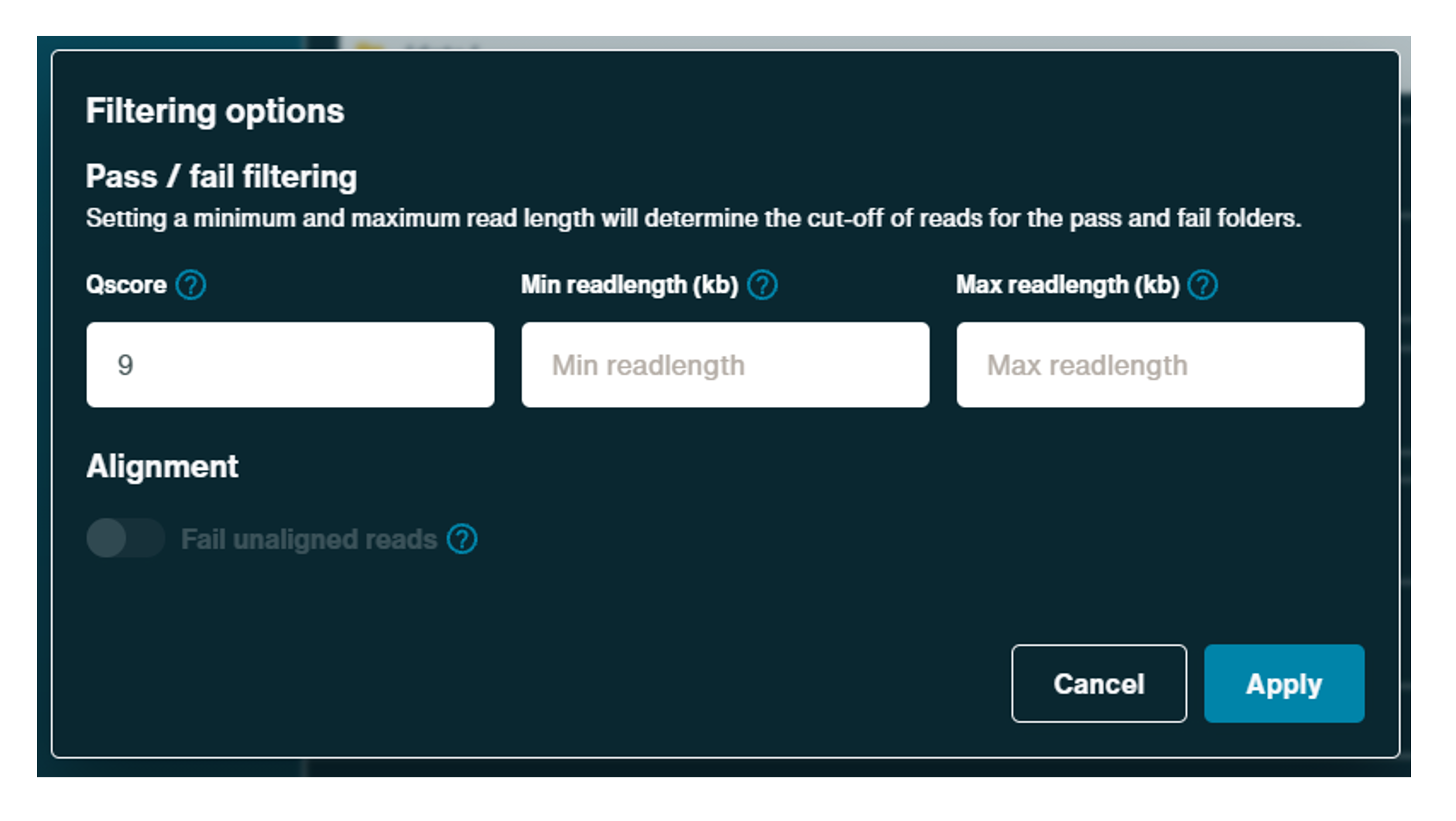
Select Continue to final review to proceed.
-
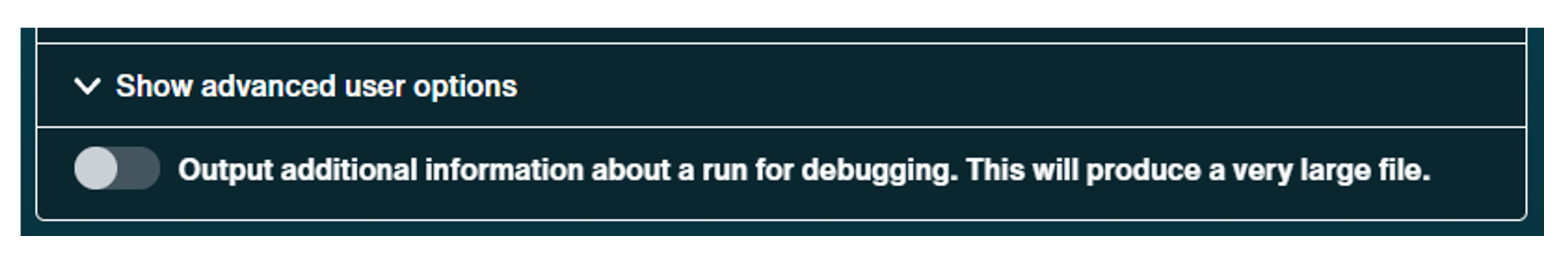
Option to turn bulk file saving either on or off.
Note: This will result in much larger file sizes due to additional information about the run, which is used for debugging.
-
Click 'Start' to run the experiment.
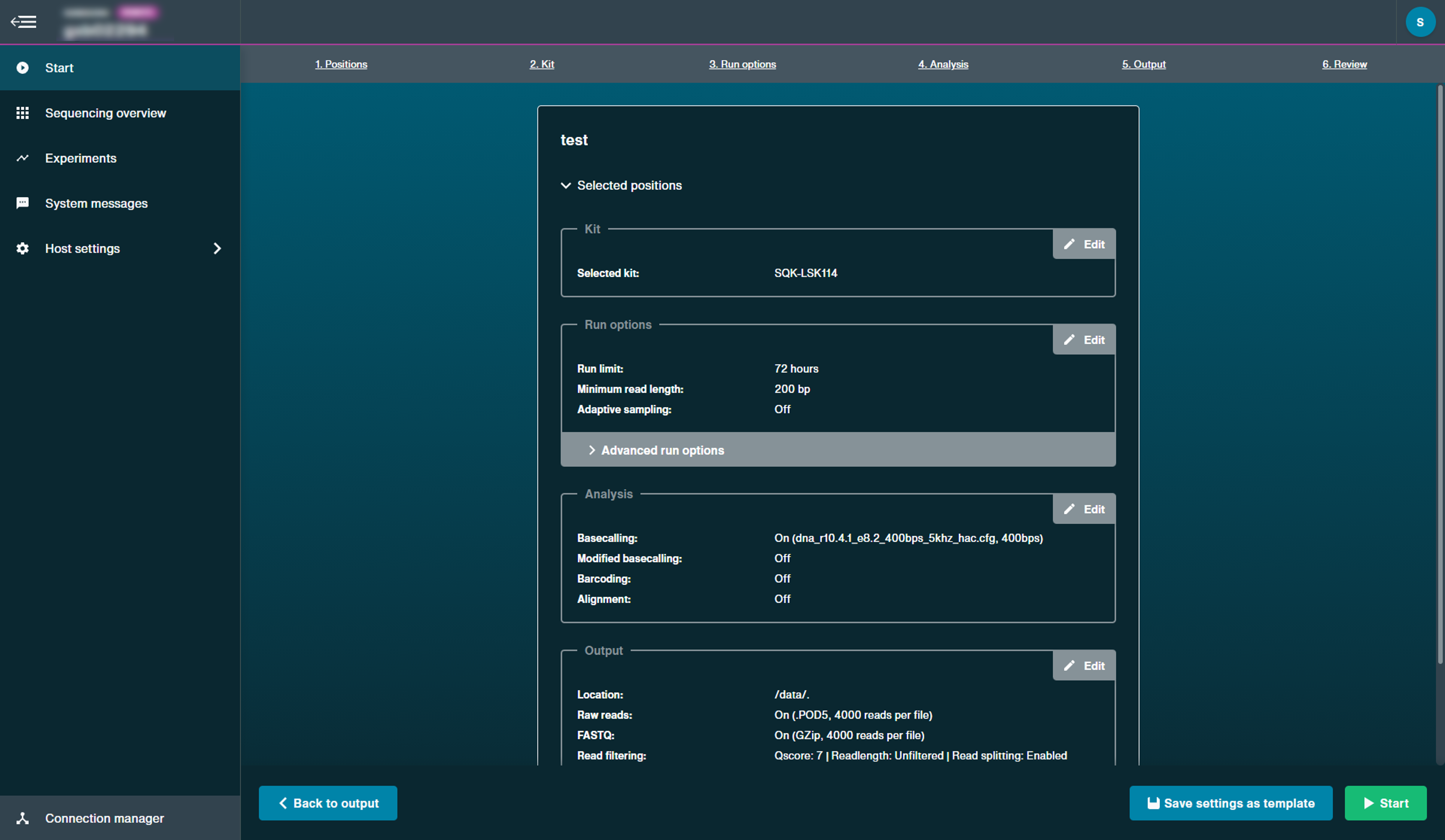
The Review page is an overview of all run options selected.
Click the Edit button to return to the set-up page and alter your parameters.
Click Advanced run options to view the additional options chosen.
-
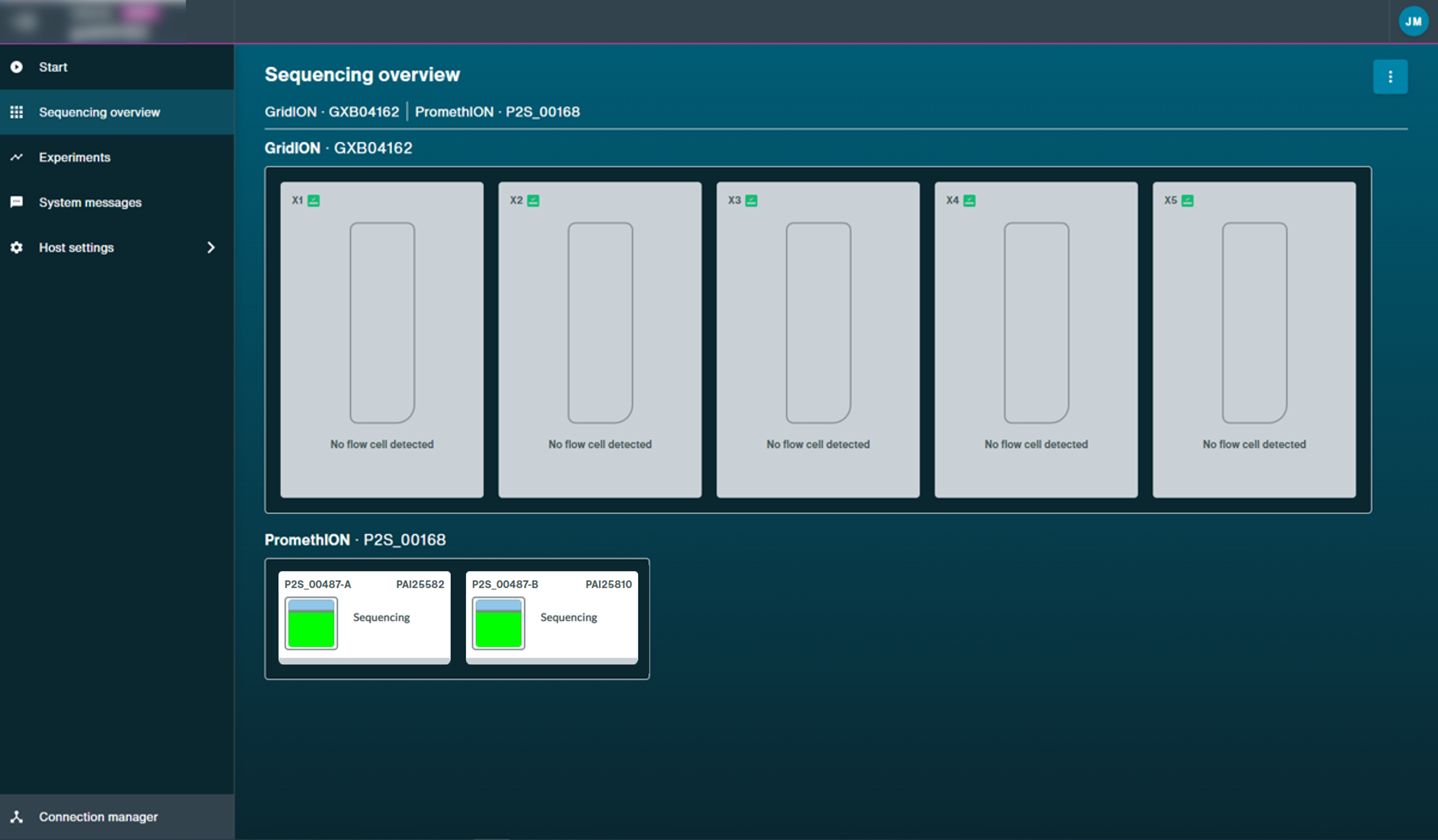
You will be automatically navigated to the Sequencing Overview when sequencing starts.
From here, you can see a progress bar below the flow cell to show the progression of the sequencing script.
In some cases, the device will take a few minutes before starting a sequencing run if an alignment reference file is used. A progress bar will show the progress before sequencing begins.
Flow cell health will be displayed after the first pore scan.
-
Click on the flow cell to open the quick view and check the number of active pores. The first pore scan should report a similar number of active pores (within 10-15%) to that reported in the flow cell check.
Below are recommended troubleshooting tips if there are unexpected differences in pore numbers:
- If there is a significant reduction in active pores in the first pore scan, restart MinKNOW.
- If the numbers are still significantly different, close down the host computer and reboot.
- When the numbers are similar to those reported at the end of the flow cell check, restart the experiment. There is no need to load any additional library after the restart.
-
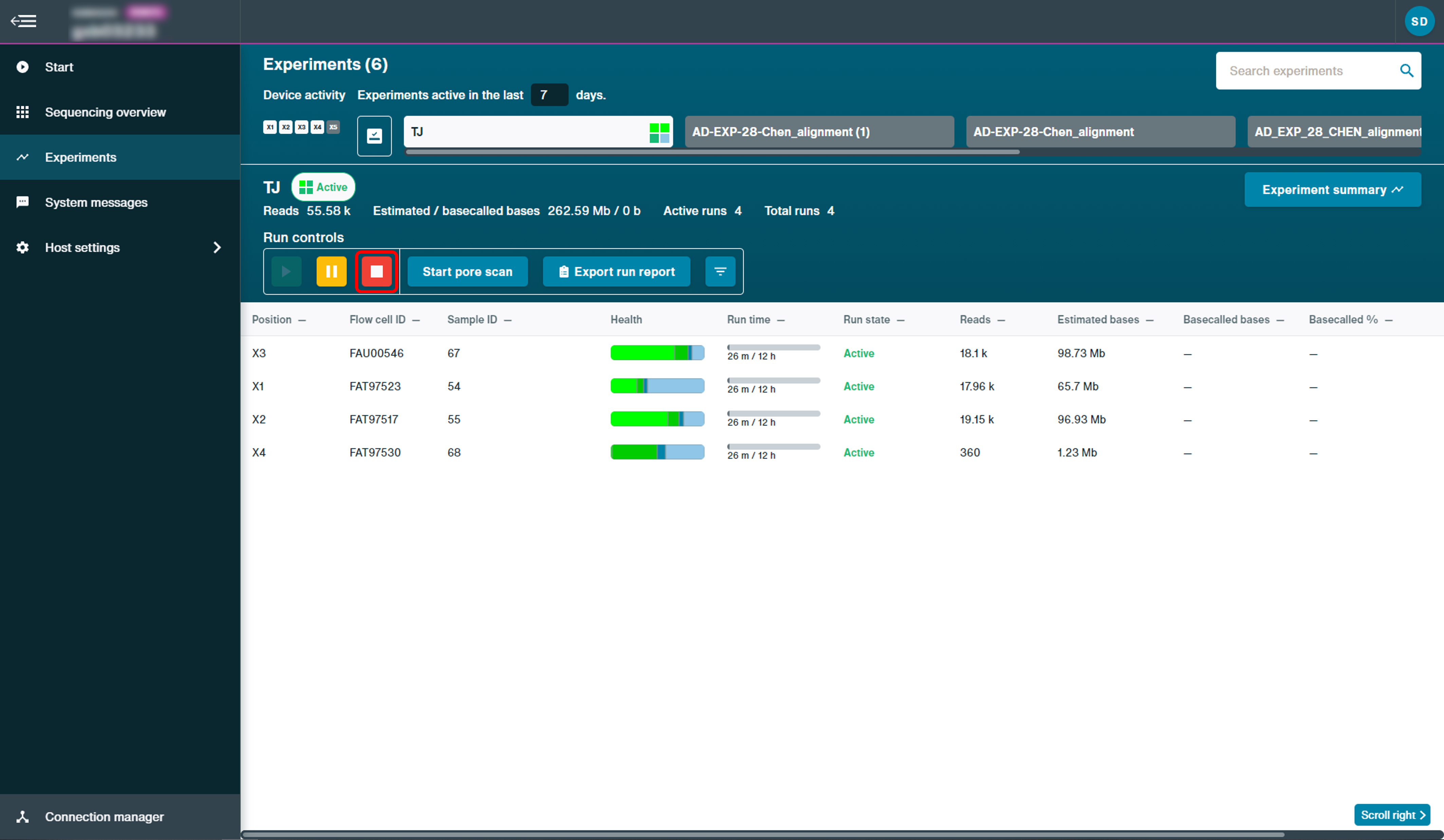
To stop the experiment during sequencing, click "Stop" using the run controls on the experiments page.
This is only necessary if you want to stop your run before the end of the set run time.
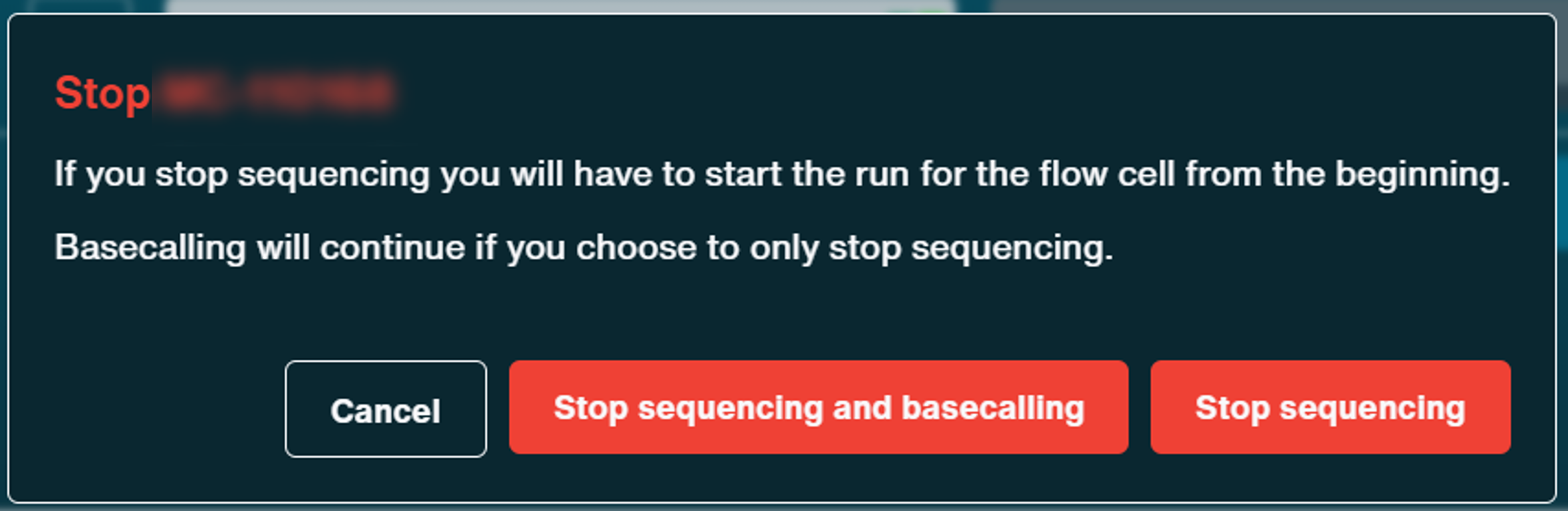
Confirm to stop experiment by clicking:
- Stop sequencing which will start catch-up basecalling
- or Stop sequencing and basecalling which will stop both sequencing and basecalling immediately.
-
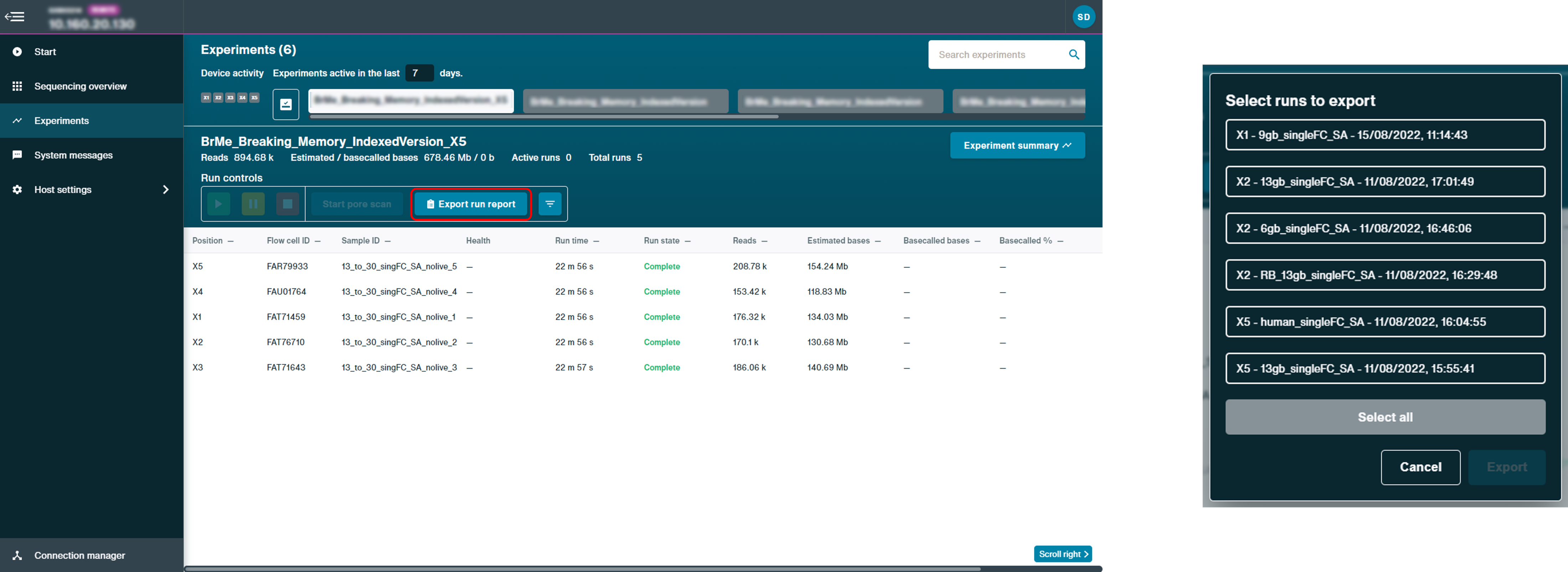
Once sequencing is complete, generate a run report.
A run report containing information about the sequencing run and performance graphs are automatically generated. To manually generate a run report, click Export run report and selecting which experiment to export to html.
For more information about the run report, please see the Run report section of this document.
A pore activity .csv file is also generated for every run.
The report and .csv files are saved to the same folder as the .fast5 and .fastq read files e.g. :\data\experiment\sample_ID\ for MinION Mk1B.