Welcome to the Nanopore Community
Order MinION devices and consumables
Visit vwr.comPCR tiling of African Swine Fever (ASF) virus (SQK-LSK109 with EXP-NBD104 or EXP-NBD114)
Version for device: MinION
Introduction to the protocol
Overview of the protocol
Overview of the protocol
-
Native Barcoding Expansion 1-12 and 13-24 features
These kits are recommended for users who:
- wish to multiplex samples to reduce price per sample
- want to optimise their sequencing experiment for throughput
- require control over read length
- are interested in utilising upstream processes such as size selection or whole genome amplification
-
Introduction to the ASFV Native Barcoding protocol
This protocol describes an efficient, low-cost method to sequence ASFV at scale. The method uses tiled PCR amplification of the virus to obtain good coverage, while enabling sample multiplexing to reduce run costs.
The kits used are the Native Barcoding Expansion 1-12 (EXP-NBD104) and 13-24 (EXP-NBD114), in conjunction with the Ligation Sequencing Kit (SQK-LSK109). There are 24 unique barcodes if using both expansion kits, allowing the user to pool up to 24 different samples in one sequencing experiment. It is highly recommended that a Lambda control experiment is completed first to become familiar with the technology.
Steps in the sequencing workflow:
Prepare for your experiment
You will need to:
- Extract your DNA, and check its length, quantity and purity The quality checks performed during the protocol are essential in ensuring experimental success
- Ensure you have your sequencing kit, the correct equipment and third-party reagents
- Download the software for acquiring and analysing your data
- Check your flow cell to ensure it has enough pores for a good sequencing run
Prepare your library
You will need to:
- Amplify the viral genome using tiled primers, purify and pool the amplicons
- Repair the DNA, and prepare the DNA ends for barcode attachment
- Ligate Native barcodes supplied in the kit to the DNA ends
- Ligate sequencing adapters supplied in the kit to the DNA ends
- Prime the flow cell, and load your DNA library into the flow cell

Sequencing
You will need to:
- Start a sequencing run using the MinKNOW software, which will collect raw data from the device and convert it into basecalled reads
- Assemble and analyse the ASF genome using bioinformatics tools of your choice (recommendations included in the "Downstream analysis" sections of the protocol)

Equipment and consumables
- Materials
-
- Ligation Sequencing Kit (SQK-LSK109)
- Flow Cell Priming Kit (EXP-FLP002)
- Native Barcoding Expansion 1-12 (EXP-NBD104) and 13-24 (EXP-NBD114) if multiplexing more than 12 samples
- ASFV-positive blood samples
- Consumables
-
- ASFV primers
- Agencourt AMPure XP beads (Beckman Coulter, A63881)
- Lysis buffer (10 mM ammonium chloride, 150 mM sodium EDTA, 10 mM sodium bicarbonate)
- 5x TEN buffer (0.05 M EDTA, 0.5 M NaCl, 20 mg/ml Proteinase K, 20% SDS, in 0.05 M Tris-HCl, pH 8.0)
- Isopropanol, 100% (Fisher, 10723124)
- NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing (NEB, E7180S or E7180L). Alternatively, you can use the NEBNext® products below:
- NEBNext FFPE Repair Mix (NEB, M6630)
- NEBNext Ultra II End repair/dA-tailing Module (NEB, E7546)
- NEBNext Quick Ligation Module (NEB, E6056)
- 1.5 ml Eppendorf DNA LoBind tubes
- 0.2 ml thin-walled PCR tubes
- Nuclease-free water (e.g. ThermoFisher, AM9937)
- Freshly prepared 70% ethanol in nuclease-free water
- Equipment
-
- Heat block or water bath set to 95°C
- Hula mixer (gentle rotator mixer)
- Magnetic rack, suitable for 1.5 ml Eppendorf tubes
- Microfuge
- Vortex mixer
- Thermal cycler
- P1000 pipette and tips
- P200 pipette and tips
- P100 pipette and tips
- P20 pipette and tips
- P10 pipette and tips
- P2 pipette and tips
- Ice bucket with ice
- Timer
- Optional Equipment
-
- Agilent Bioanalyzer (or equivalent)
- Qubit fluorometer (or equivalent for QC check)
- Eppendorf 5424 centrifuge (or equivalent)
-
NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing
For customers new to nanopore sequencing, we recommend buying the NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing (catalogue number E7180S or E7180L), which contains all the NEB reagents needed for use with the Ligation Sequencing Kit.
Please note, for our amplicon protocols, NEBNext FFPE DNA Repair Mix and NEBNext FFPE DNA Repair Buffer are not required.
-
Ligation Sequencing Kit (SQK-LSK109) contents

Name Acronym Cap colour No. of vials Fill volume per vial (µl) DNA CS DCS Yellow 1 50 Adapter Mix AMX Green 1 40 Ligation Buffer LNB Clear 1 200 L Fragment Buffer LFB White cap, orange stripe on label 2 1,800 S Fragment Buffer SFB Grey 2 1,800 Sequencing Buffer SQB Red 2 300 Elution Buffer EB Black 1 200 Loading Beads LB Pink 1 360 -
Flow Cell Priming Kit (EXP-FLP002) contents
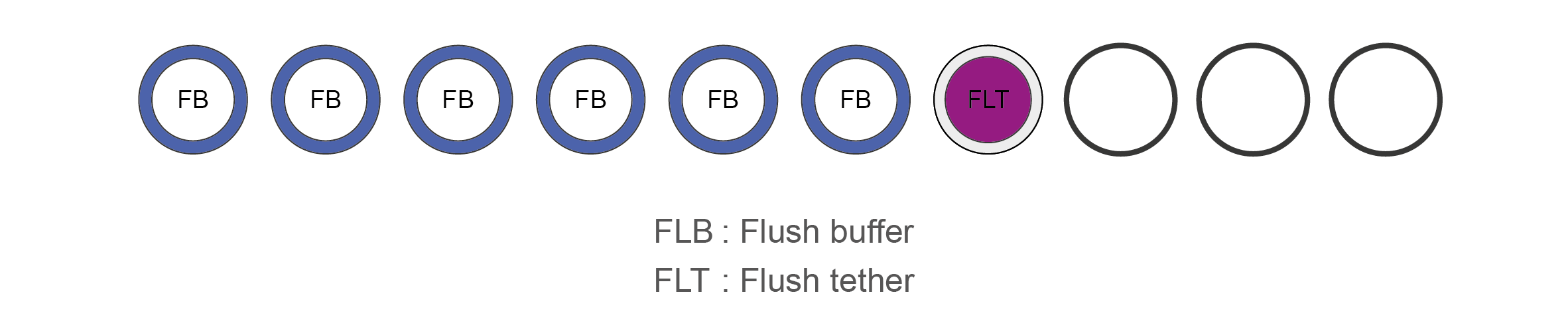
Name Acronym Cap colour No. of vial Fill volume per vial (μl) Flush Buffer FB Blue 6 1,170 Flush Tether FLT Purple 1 200 -
Native Barcoding Expansion 1-12 (EXP-NBD104) and 13-24 (EXP-NBD114) contents
EXP-NBD104 kit contents

Name Acronym Cap colour No. of vials Fill volume per vial (μl) Native Barcode 01-12 NB01-12 White 12 20 Adapter Mix II AMII Green 1 40
EXP-NBD114 kit contents

Name Acronym Cap colour No. of vials Fill volume per vial (μl) Native Barcode 13-24 NB13-24 White 12 20 Adapter Mix II AMII Green 1 40 -
Native barcode sequences
The native barcode sequences are the reverse complement of the corresponding barcode sequence in other kits. 24 unique barcodes are available in the Native Barcoding Expansion 1-12 and 13-24 (EXP-NBD104 and EXP-NBD114).
Native Barcoding Expansion 1-12 and 13-24 (EXP-NBD104 and EXP-NBD114)
Component Forward sequence Reverse sequence NB01 CACAAAGACACCGACAACTTTCTT AAGAAAGTTGTCGGTGTCTTTGTG NB02 ACAGACGACTACAAACGGAATCGA TCGATTCCGTTTGTAGTCGTCTGT NB03 CCTGGTAACTGGGACACAAGACTC GAGTCTTGTGTCCCAGTTACCAGG NB04 TAGGGAAACACGATAGAATCCGAA TTCGGATTCTATCGTGTTTCCCTA NB05 AAGGTTACACAAACCCTGGACAAG CTTGTCCAGGGTTTGTGTAACCTT NB06 GACTACTTTCTGCCTTTGCGAGAA TTCTCGCAAAGGCAGAAAGTAGTC NB07 AAGGATTCATTCCCACGGTAACAC GTGTTACCGTGGGAATGAATCCTT NB08 ACGTAACTTGGTTTGTTCCCTGAA TTCAGGGAACAAACCAAGTTACGT NB09 AACCAAGACTCGCTGTGCCTAGTT AACTAGGCACAGCGAGTCTTGGTT NB10 GAGAGGACAAAGGTTTCAACGCTT AAGCGTTGAAACCTTTGTCCTCTC NB11 TCCATTCCCTCCGATAGATGAAAC GTTTCATCTATCGGAGGGAATGGA NB12 TCCGATTCTGCTTCTTTCTACCTG CAGGTAGAAAGAAGCAGAATCGGA NB13 AGAACGACTTCCATACTCGTGTGA TCACACGAGTATGGAAGTCGTTCT NB14 AACGAGTCTCTTGGGACCCATAGA TCTATGGGTCCCAAGAGACTCGTT NB15 AGGTCTACCTCGCTAACACCACTG CAGTGGTGTTAGCGAGGTAGACCT NB16 CGTCAACTGACAGTGGTTCGTACT AGTACGAACCACTGTCAGTTGACG NB17 ACCCTCCAGGAAAGTACCTCTGAT ATCAGAGGTACTTTCCTGGAGGGT NB18 CCAAACCCAACAACCTAGATAGGC GCCTATCTAGGTTGTTGGGTTTGG NB19 GTTCCTCGTGCAGTGTCAAGAGAT ATCTCTTGACACTGCACGAGGAAC NB20 TTGCGTCCTGTTACGAGAACTCAT ATGAGTTCTCGTAACAGGACGCAA NB21 GAGCCTCTCATTGTCCGTTCTCTA TAGAGAACGGACAATGAGAGGCTC NB22 ACCACTGCCATGTATCAAAGTACG CGTACTTTGATACATGGCAGTGGT NB23 CTTACTACCCAGTGAACCTCCTCG CGAGGAGGTTCACTGGGTAGTAAG NB24 GCATAGTTCTGCATGATGGGTTAG CTAACCCATCATGCAGAACTATGC -
ASFV primer sequences
ASFV primer sequences described in the protocol are subject to change. Any updates can be found at ASFV Lilo GitHub page.
Amplicon - primer pair Forward sequence Reverse sequence ASFV1 GGCGTTCATTTCACAAGATGC ACGGCATCTAAGCAGCTCAATG ASFV2 CAGGCCGATATATCATTTCATCAATATTCA ACCCAAAGCCCTGGAATCCTTA ASFV3 GCAAACCAAGTGACTCACCCTC ATTGTATGACGTCGGGGCAGAT ASFV4 ACCTAGTAAAAGTCCTAGAAAAACCTTCA CGCCATTGTTTTACACAGTCGC ASFV5 TCGAGATTTTATTATTTGGATGCATCATCA GGACTGATGAAAGCCGTGAA ASFV6 TCCACGCGGTACTTGGCTCC AGGCCTCGTTGGTGGAAAGGA ASFV7 AGGGCTGATGCAAATCTCTTTTTCA TCTCCGATTTTCGCATGCCAAA ASFV8 AGTTTGCAAAGAGCCTAAAGATAGACT AGCGTGGAACTGTAGATGACGA ASFV9 TGCAGAAACCGCAGATGAATGT ATAGGATTAGATGCGACGCCCA ASFV10 GCATGTAGAGAGGTTTTGGTAGTCA GGAAACAGCTGGAGAGTTGTGG ASFV11 TGGTTTTGAAATAAAATGCCTTCTACGG GGAATGCATGGACGAAGAAGCA ASFV11 (alt) CTATGGGATGGGAAGAGTGGTCAA CGTCAACCGCCGCATTAGC ASFV12 TCCTTGGGAGTTACAGCGAAGA AATGAAATCATTCGCGGCGAGT ASFV13 CAGACATTGGCAGTGATGGCTA GAAATGCCGGGCCTTCTACAAA ASFV14 GCTACTCCCCCAAATATCACATATAATTGT TTTTTCGTGTTGCTGTTCGGGA ASFV15 GGATGGCACCCTTCTCACAATC TGCGTATGACCCGATGTTGTTG ASFV16 GTCGACTTCACAGGAACAACGG ACCCGCTTTACACAAAACACGT ASFV17 TGGAATTTCCTGACGTGGCAAA GCAACCGCTATTCCAAACAGGA ASFV18 AGTTGTTGTCCTAGACCGTGGCA TGAAAAGGAGGGCACGATCC ASFV19 CCCGTATGCGGGCGTACTTT TGGCCTCTTCTTTCCCCCGA ASFV20 GGCCGCAACATTTGTGTCAAAG GCTCGCGAACAAATTACTCCCA ASFV21 GAATGGCAGCGATGATCTCAGG TGCAGGGCAAGGGTATACTGAA ASFV22 TGGCGTCGTTTAACAGCTTGAT GCTGGATGGCAAATCGGTTGTA ASFV23 AGGCGTGAAAATTCTTCTTCAAACA AGACGTTTTAAGCTGCATGGCA ASFV24 GGCAGCAGGATCTTAAAACCGG TGCATAATGCCCAGCTTTTCGT ASFV25 GCTGTTTAAGCGTTTCAAGCTGA CTCCGCGGGGAACATTGTTTTA ASFV26 CCCTGGGAGGAGTCATCATGAA GGTCATTGACTTTGGAAGCGCT ASFV27 ACTGTCTGCTAGACTCCCAGGA CCCAAGAGGAGGAATGGTTTGC ASFV28 GCCCCCTAGCGTCACCGAAT CCAAGCCTGCTGCGAAGCTC ASFV29 GACGCAATTTCGGCTGTTTTTAAAA GACTTGGTCTCCGGCTCAAAAG ASFV30 GTTGGGGTGTTGGAGCGAATAA TTCTGCTTACGGACGATGCAAC ASFV31 TAGTTGTGAAGCGTTCTCGGGT GAGCACATGTTACTCGCCACTC ASFV32 GGACTTCTTATGCTCAGATGGGC ACTGCTGCAGGCGTTAAACATT




Computer requirements and software
Computer requirements and software
-
MinION Mk1B IT requirements
Sequencing on a MinION Mk1B requires a high-spec computer or laptop to keep up with the rate of data acquisition. For more information, refer to the MinION Mk1B IT requirements document.
-
MinION Mk1C IT requirements
The MinION Mk1C contains fully-integrated compute and screen, removing the need for any accessories to generate and analyse nanopore data. For more information refer to the MinION Mk1C IT requirements document.
-
MinION Mk1D IT requirements
Sequencing on a MinION Mk1D requires a high-spec computer or laptop to keep up with the rate of data acquisition. For more information, refer to the MinION Mk1D IT requirements document.
-
Software for nanopore sequencing
MinKNOW
The MinKNOW software controls the nanopore sequencing device, collects sequencing data and basecalls in real time. You will be using MinKNOW for every sequencing experiment to sequence, basecall and demultiplex if your samples were barcoded.
For instructions on how to run the MinKNOW software, please refer to the MinKNOW protocol.
EPI2ME (optional)
The EPI2ME cloud-based platform performs further analysis of basecalled data, for example alignment to the Lambda genome, barcoding, or taxonomic classification. You will use the EPI2ME platform only if you would like further analysis of your data post-basecalling.
For instructions on how to create an EPI2ME account and install the EPI2ME Desktop Agent, please refer to this link.
-
Check your flow cell
We highly recommend that you check the number of pores in your flow cell prior to starting a sequencing experiment. This should be done within 12 weeks of purchasing for MinION/GridION/PromethION or within four weeks of purchasing Flongle Flow Cells. Oxford Nanopore Technologies will replace any flow cell with fewer than the number of pores in the table below, when the result is reported within two days of performing the flow cell check, and when the storage recommendations have been followed. To do the flow cell check, please follow the instructions in the Flow Cell Check document.
Flow cell Minimum number of active pores covered by warranty Flongle Flow Cell 50 MinION/GridION Flow Cell 800 PromethION Flow Cell 5000
Library preparation
DNA extraction
DNA extraction
- Materials
-
- ASFV-positive blood samples
- Consumables
-
- Lysis buffer (10 mM ammonium chloride, 150 mM sodium EDTA, 10 mM sodium bicarbonate)
- 5x TEN buffer (0.05 M EDTA, 0.5 M NaCl, 20 mg/ml Proteinase K, 20% SDS, in 0.05 M Tris-HCl, pH 8.0)
- Freshly prepared 70% ethanol in nuclease-free water
- Isopropanol, 100% (Fisher, 10723124)
- Nuclease-free water
- 1.5 ml Eppendorf DNA LoBind tubes
- Equipment
-
- Microfuge
- Heat block or water bath set to 95°C
- Hula mixer (gentle rotator mixer)
- P1000 pipette and tips
- P200 pipette and tips
- P100 pipette and tips
- P20 pipette and tips
-
The blood samples used will have been tested for ASFV by PCR prior to DNA extraction. Individual blood samples are preferred; however this protocol can also work with pooled blood samples from multiple animals.
-
Transfer 50 μl of whole blood to a 1.5 ml Eppendorf DNA LoBind tube.
-
Add 1 ml of lysis buffer to each sample.
-
Incubate on a Hula mixer (rotator mixer) for 1 minute at room temperature.
-
Remove the samples from the Hula mixer and incubate at room temperature for 30 minutes.
-
Centrifuge at 7,500 rpm for 5 minutes. You should see a pellet form at the bottom of the tube.
-
Without disturbing the pellet, remove and discard the supernatant.
-
Add 700 μl of 5X TEN buffer to each sample.
-
Incubate on a Hula mixer (rotator mixer) for 1 minute at room temperature.
-
Remove the samples from the Hula mixer and incubate at room temperature for 30 minutes.
-
Incubate the samples at 95°C for 5 minutes in a heat block or water bath.
-
Centrifuge at 7,500 rpm for 5 minutes. You should see a pellet form at the bottom of the tube.
-
Without disturbing the pellet, remove and discard the supernatant.
-
Resuspend each pellet in 700 μl 100% isopropanol.
-
Incubate on a Hula mixer (rotator mixer) for 1 minute at room temperature.
-
Remove the samples from the Hula mixer and incubate at room temperature for 30 minutes.
-
Prepare sufficient fresh 70% ethanol in nuclease-free water.
-
Centrifuge the samples at 14,500 rpm for 15 minutes. You should see a white pellet form at the bottom of the tube.
-
Without disturbing the pellet, remove and discard the supernatant.
-
Wash the pellet by adding 500 μl of freshly-prepared 70% ethanol and pipetting up and down.
-
Centrifuge the samples at 11,500 rpm for 5 minutes.
-
Without disturbing the pellet, remove and discard the supernatant.
-
Allow the pellet to dry for ~30 seconds, ensuring there is no ethanol left in the sample. However, do not dry the pellet to the point of cracking.
-
Resuspend the pellet in 30 μl nuclease-free water. If it is difficult to resuspend in this volume, add more nuclease-free water.
Tiled DNA amplification and clean-up
Tiled DNA amplification and clean-up
- Materials
-
- Extracted ASFV DNA
- Consumables
-
- ASFV primers
- VeriFi HS Mix (PCRBIO)
- Nuclease-free water
- Agencourt AMPure XP Beads (Beckman Coulter™, A63881)
- Freshly prepared 70% ethanol in nuclease-free water
- 0.2 ml thin-walled PCR tubes
- 1.5 ml Eppendorf DNA LoBind tubes
- Qubit™ Assay Tubes (Invitrogen, Q32856)
- Qubit dsDNA BR Assay Kit (Invitrogen, Q32850)
- Equipment
-
- Microfuge
- Thermal cycler and/or heating block
- Hula mixer (gentle rotator mixer)
- Magnetic rack suitable for 0.2 ml PCR tubes
- P1000 pipette and tips
- P200 pipette and tips
- P100 pipette and tips
- P20 pipette and tips
- P2 pipette and tips
- Qubit fluorometer (or equivalent for QC check)
-
Prepare the primers according to the manufacturer's instructions.
-
Pool the tiled primer pairs in Eppendorf or PCR tubes following these proportions. The final stock concentration should be 100 μM.
Odd primer pool:
Primer pair Concentration 3 0.75X 5 2X 7 1X 9 1X 11 1X 11 alt 1X 13 1X 15 1X 17 1.5X 19 1X 21 1X 23 1X 25 2X 27 1X 29 1X 31 0.5X Even primer pool:
Primer pair Concentration 2 1X 4 2X 6 0.5X 8 1.5X 10 2X 12 2X 14 2.5X 16 1X 18 1.5X 20 1X 22 1.5X 24 1.5X 26 1.5X 28 0.75X 30 1.5X 32 1X Primer one:
Primer number Concentration 1 1X Note: Due to the proximity to the telomeric sequence, primer 1 has a shorter amplicon length. The primer does not perform optimally in when pooled with others, therefore it is prepared separately.
-
Optional ActionTo conserve the DNA samples, dilute 1:10 using nuclease-free water. Unused sample should be stored at –20°C.
-
Immediately before setting up the PCR, dilute each primer pool 1:10 in nuclease-free water to make a 10 μM working stock.
-
For each sample, prepare one reaction corresponding to each primer pool in 0.2 ml PCR tubes:
Note: For each sample you will have three reactions – odd primer pool, even primer pool, and primer 1 pool.
Between each addition, pipette mix 10-20 times.
Reagent Volume Nuclease-free water 9 µl ASFV DNA (1:10 dilution) 2 µl Primer pool (1:10 dilution) 1.5 µl VeriFi HS Mix (PCRBIO) 12.5 µl Total 25 µl Note: we recommend to make a PCR master mix, either for individual primer pools for multiple samples or one for the three primers sets.
-
Mix well by pipetting and spin down.
-
Incubate in a thermocycler using the following program:
Step Temperature Time Cycles Initial denaturation 98°C 1 min 1 Denaturation
Annealing
Extension98°C
60°C
72°C15 sec
15 sec
4 min 40 sec
40Final extension 72°C 5 min 1 Hold 10°C ∞ -
Resuspend the AMPure XP beads by vortexing.
-
Add 10 µl of resuspended AMPure XP beads to each tube and mix by gently pipetting.
-
Incubate for 10 minutes at room temperature.
-
Prepare 50 ml of fresh 70% ethanol in nuclease-free water.
-
Spin down the tubes and pellet the beads on a magnet for 5 minutes. Keep the tubes on the magnet until the eluate is clear and colourless, and pipette off the supernatant.
-
Keep the tubes on the magnet and wash the beads in each well with 200 µl of freshly prepared 70% ethanol without disturbing the pellet. Remove the ethanol using a pipette and discard.
-
Repeat the previous step.
-
Spin down and place the tubes back on the magnet. Pipette off any residual ethanol. Allow to dry for ~30 seconds, but do not dry the pellet to the point of cracking.
-
Remove the tubes from the magnetic rack and resuspend each pellet in 15 µl nuclease-free water. Incubate for 2 minutes at room temperature.
-
Pellet the beads on a magnet until the eluate is clear and colourless.
-
Remove and retain 15 µl of eluate containing the DNA for each sample, in its three respective primer pools, into a clean tube.
Note: At this point you should still have three tubes for each DNA sample.
-
Quantify 1 μl of each cleaned PCR product using a Qubit ds DNA BR assay.
-
Using the Qubit reads, pool each sample by quantity in the following proportions:
Primer pool Proportion Pool 1 48% Pool 2 50% Amplicon 1 2%
DNA repair and end-prep
DNA repair and end-prep
- Materials
-
- Pooled ASFV amplicons
- Consumables
-
- 0.2 ml thin-walled PCR tubes
- Nuclease-free water (e.g. ThermoFisher, AM9937)
- NEBNext FFPE DNA Repair Mix (NEB, M6630)
- NEBNext® Ultra II End Repair / dA-tailing Module (NEB, E7546)
- Agencourt AMPure XP Beads (Beckman Coulter™, A63881)
- Freshly prepared 70% ethanol in nuclease-free water
- 1.5 ml Eppendorf DNA LoBind tubes
- Equipment
-
- P1000 pipette and tips
- P100 pipette and tips
- P10 pipette and tips
- Thermal cycler
- Microfuge
- Hula mixer (gentle rotator mixer)
- Magnetic rack
- Ice bucket with ice
-
Prepare the NEBNext FFPE DNA Repair Mix and NEBNext Ultra II End Repair / dA-tailing Module reagents in accordance with manufacturer’s instructions, and place on ice.
For optimal performance, NEB recommend the following:
Thaw all reagents on ice.
Flick and/or invert the reagent tubes to ensure they are well mixed.
Note: Do not vortex the FFPE DNA Repair Mix or Ultra II End Prep Enzyme Mix.Always spin down tubes before opening for the first time each day.
The Ultra II End Prep Buffer and FFPE DNA Repair Buffer may have a little precipitate. Allow the mixture to come to room temperature and pipette the buffer up and down several times to break up the precipitate, followed by vortexing the tube for 30 seconds to solubilise any precipitate.
Note: It is important the buffers are mixed well by vortexing.The FFPE DNA Repair Buffer may have a yellow tinge and is fine to use if yellow.
-
Prepare the DNA in nuclease-free water.
- Transfer 700 ng of amplicon DNA into a 1.5 ml Eppendorf DNA LoBind tube
- Adjust the volume to 48 μl with nuclease-free water
- Mix thoroughly by flicking the tube
- Spin down briefly in a microfuge
-
In a 0.2 ml thin-walled PCR tube, mix the following:
Between each addition, pipette mix 10-20 times
Reagent Volume DNA 48 µl NEBNext FFPE DNA Repair Buffer 3.5 µl Ultra II End-prep reaction buffer 3.5 µl Ultra II End-prep enzyme mix 3 µl NEBNext FFPE DNA Repair Mix 2 µl Total 60 µl -
Mix well by pipetting and spin down.
-
Using a thermal cycler, incubate at 20°C for 5 minutes and 65°C for 5 minutes.
-
Transfer the DNA sample to a clean 1.5 ml Eppendorf DNA LoBind tube.
-
Resuspend the AMPure XP beads by vortexing.
-
Add 60 µl of resuspended AMPure XP beads to the end-prep reaction and mix by flicking the tube.
-
Incubate on a Hula mixer (rotator mixer) for 5 minutes at room temperature.
-
Prepare 500 μl of fresh 70% ethanol in nuclease-free water.
-
Spin down the sample and pellet on a magnet until supernatant is clear and colourless. Keep the tube on the magnet, and pipette off the supernatant.
-
Keep the tube on the magnet and wash the beads with 200 µl of freshly prepared 70% ethanol without disturbing the pellet. Remove the ethanol using a pipette and discard.
-
Repeat the previous step.
-
Spin down and place the tube back on the magnet. Pipette off any residual ethanol. Allow to dry for ~30 seconds, but do not dry the pellet to the point of cracking.
-
Remove the tube from the magnetic rack and resuspend the pellet in 25 µl nuclease-free water. Spin down and incubate for 5 minutes at room temperature.
-
Pellet the beads on a magnet until the eluate is clear and colourless, for at least 1 minute.
-
Remove and retain 25 µl of eluate into a clean 1.5 ml Eppendorf DNA LoBind tube.
-
Optional ActionQuantify 1 µl of eluted sample using a Qubit fluorometer.
Native barcode ligation
- Materials
-
- Native Barcoding Expansion 1-12 (EXP-NBD104) and 13-24 (EXP-NBD114) if multiplexing more than 12 samples
- Consumables
-
- Freshly prepared 70% ethanol in nuclease-free water
- 1.5 ml Eppendorf DNA LoBind tubes
- Nuclease-free water (e.g. ThermoFisher, AM9937)
- Agencourt AMPure XP beads (Beckman Coulter, A63881)
- NEB Blunt/TA Ligase Master Mix (NEB, M0367)
- Equipment
-
- Magnetic rack, suitable for 1.5 ml Eppendorf tubes
- Hula mixer (gentle rotator mixer)
- Vortex mixer
- Ice bucket with ice
- Microfuge
- P1000 pipette and tips
- P100 pipette and tips
- P10 pipette and tips
- Optional Equipment
-
- Qubit fluorometer (or equivalent for QC check)
-
Select a unique barcode for every sample to be run together on the same flow cell, from the provided 24 barcodes. Up to 24 samples can be barcoded and combined in one experiment.
-
Thaw the native barcodes at room temperature. Use one barcode per sample. Individually mix the barcodes by pipetting, spin down, and place them on ice.
-
Dilute 500 ng of each end-prepped sample to be barcoded to 22.5 µl in nuclease-free water.
-
Add the reagents in the order given below, mixing by flicking the tube between each sequential addition:
Reagent Volume 500 ng end-prepped DNA 22.5 µl Native Barcode 2.5 µl Blunt/TA Ligase Master Mix 25 µl Total 50 µl -
Mix by pipetting up and down 10-20 times and spin down.
-
Incubate the reaction for 10 minutes at room temperature.
-
Resuspend the AMPure XP beads by vortexing.
-
Add 50 µl of resuspended AMPure XP beads to the reaction and mix by pipetting.
-
Incubate on a Hula mixer (rotator mixer) for 5 minutes at room temperature.
-
Prepare 500 μl of fresh 70% ethanol in nuclease-free water.
-
Spin down the sample and pellet on a magnet. Keep the tube on the magnet, and pipette off the supernatant when clear and colourless.
-
Keep the tube on the magnet and wash the beads with 200 µl of freshly prepared 70% ethanol without disturbing the pellet. Remove the ethanol using a pipette and discard.
-
Repeat the previous step.
-
Spin down and place the tube back on the magnet. Pipette off any residual ethanol. Allow to dry for ~30 seconds, but do not dry the pellet to the point of cracking.
-
Remove the tube from the magnetic rack and resuspend the pellet in 26 µl nuclease-free water. Incubate for 2 minutes at room temperature.
-
Pellet the beads on a magnet until the eluate is clear and colourless.
-
Remove and retain 26 µl of eluate containing the DNA library into a clean 1.5 ml Eppendorf DNA LoBind tube.
Dispose of the pelleted beads
-
Quantify 1 µl of eluted sample using a Qubit fluorometer.
-
Pool equimolar amounts of each barcoded sample into a 1.5 ml Eppendorf DNA LoBind tube, ensuring that sufficient sample is combined to produce a pooled sample of 700 ng total.
-
Quantify 1 µl of pooled and barcoded DNA using a Qubit fluorometer.
-
Dilute 700 ng pooled sample to 65 µl in nuclease-free water.
-
Optional ActionIf 700 ng of pooled sample exceeds 65 µl in volume, perform an AMPure clean-up with 2.5x Agencourt AMPure XP beads to pooled sample volume, eluting in 65 µl of nuclease-free water.
Adapter ligation and clean-up
Adapter ligation and clean-up
- Materials
-
- Long Fragment Buffer (LFB)
- Elution Buffer (EB)
- Adapter Mix II (AMII)
- Consumables
-
- NEBNext® Quick Ligation Module (NEB, E6056)
- NEBNext® Quick Ligation Reaction Buffer (NEB, B6058)
- Agencourt AMPure XP beads (Beckman Coulter™, A63881)
- 1.5 ml Eppendorf DNA LoBind tubes
- Equipment
-
- Microfuge
- Magnetic rack
- Vortex mixer
- Hula mixer (gentle rotator mixer)
- Optional Equipment
-
- Qubit fluorometer (or equivalent for QC check)
-
Adapter Mix II Expansion use
Protocols that use the Native Barcoding Expansions require 5 μl of AMII per reaction. Native Barcoding Expansions EXP-NBD104/NBD114 contain sufficient AMII for 6 reactions (or 12 reactions when sequencing on Flongle). This assumes that all barcodes are used in one sequencing run.
The Adapter Mix II expansion provides additional AMII for customers who are running subsets of barcodes, and allows a further 12 reactions (24 on Flongle).
-
Thaw the Elution Buffer (EB) and NEBNext Quick Ligation Reaction Buffer (5x) at room temperature, mix by vortexing, spin down and place on ice. Check the contents of each tube are clear of any precipitate.
-
Spin down the T4 Ligase and the Adapter Mix II (AMII), and place on ice.
-
Thaw one tube of Long Fragment Buffer (LFB) at room temperature and mix by vortexing, then spin down and place on ice.
-
Taking the pooled and barcoded DNA, perform adapter ligation as follows, mixing by flicking the tube between each sequential addition.
Reagent Volume 700 ng pooled barcoded sample 65 µl Adapter Mix II (AMII) 5 µl NEBNext Quick Ligation Reaction Buffer (5X) 20 µl Quick T4 DNA Ligase 10 µl Total 100 µl -
Ensure the components are thoroughly mixed by pipetting, and spin down.
-
Incubate the reaction for 10 minutes at room temperature.
-
Resuspend the AMPure XP beads by vortexing.
-
Add 40 µl of resuspended AMPure XP beads to the reaction and mix by pipetting.
-
Incubate on a Hula mixer (rotator mixer) for 5 minutes at room temperature.
-
Place on a magnetic rack, allow beads to pellet and pipette off supernatant.
-
Wash the beads by adding 250 μl Long Fragment Buffer (LFB). Flick the beads to resuspend, spin down, then return the tube to the magnetic rack and allow the beads to pellet. Remove the supernatant using a pipette and discard.
-
Repeat the previous step.
-
Spin down and place the tube back on the magnet. Pipette off any residual supernatant. Allow to dry for ~30 seconds, but do not dry the pellet to the point of cracking.
-
Remove the tube from the magnetic rack and resuspend the pellet in 15 µl Elution Buffer (EB). Spin down and incubate for 10 minutes at 37°C to improve the recovery of long fragments.
-
Pellet the beads on a magnet until the eluate is clear and colourless, for at least 1 minute.
-
Remove and retain 15 µl of eluate containing the DNA library into a clean 1.5 ml Eppendorf DNA LoBind tube.
Dispose of the pelleted beads
-
Quantify 1 µl of adapter ligated and barcoded DNA using a Qubit fluorometer - recovery aim ~430 ng.
-
Optional ActionIf quantities allow, the library may be diluted in Elution Buffer (EB) for splitting across multiple flow cells.
Additional buffer for doing this can be found in the Sequencing Auxiliary Vials expansion (EXP-AUX001), available to purchase separately. This expansion also contains additional vials of Sequencing Buffer (SQB) and Loading Beads (LB), required for loading the libraries onto flow cells.
Priming and loading the SpotON flow cell
Priming and loading the SpotON flow cell
- Materials
-
- Flow Cell Priming Kit (EXP-FLP002)
- Loading Beads (LB)
- Sequencing Buffer (SQB)
- Consumables
-
- 1.5 ml Eppendorf DNA LoBind tubes
- Nuclease-free water (e.g. ThermoFisher, AM9937)
- Equipment
-
- MinION device
- SpotON Flow Cell
- P1000 pipette and tips
- P100 pipette and tips
- P20 pipette and tips
- P10 pipette and tips
- MinION and GridION Flow Cell Light Shield
-
Thaw the Sequencing Buffer (SQB), Loading Beads (LB), Flush Tether (FLT) and one tube of Flush Buffer (FB) at room temperature before mixing the reagents by vortexing, and spin down at room temperature.
-
To prepare the flow cell priming mix, add 30 µl of thawed and mixed Flush Tether (FLT) directly to the tube of thawed and mixed Flush Buffer (FB), and mix by vortexing at room temperature.
-
Open the MinION device lid and slide the flow cell under the clip.
Press down firmly on the flow cell to ensure correct thermal and electrical contact.


-
Optional ActionComplete a flow cell check to assess the number of pores available before loading the library.
This step can be omitted if the flow cell has been checked previously.
See the flow cell check instructions in the MinKNOW protocol for more information.
-
Slide the flow cell priming port cover clockwise to open the priming port.

-
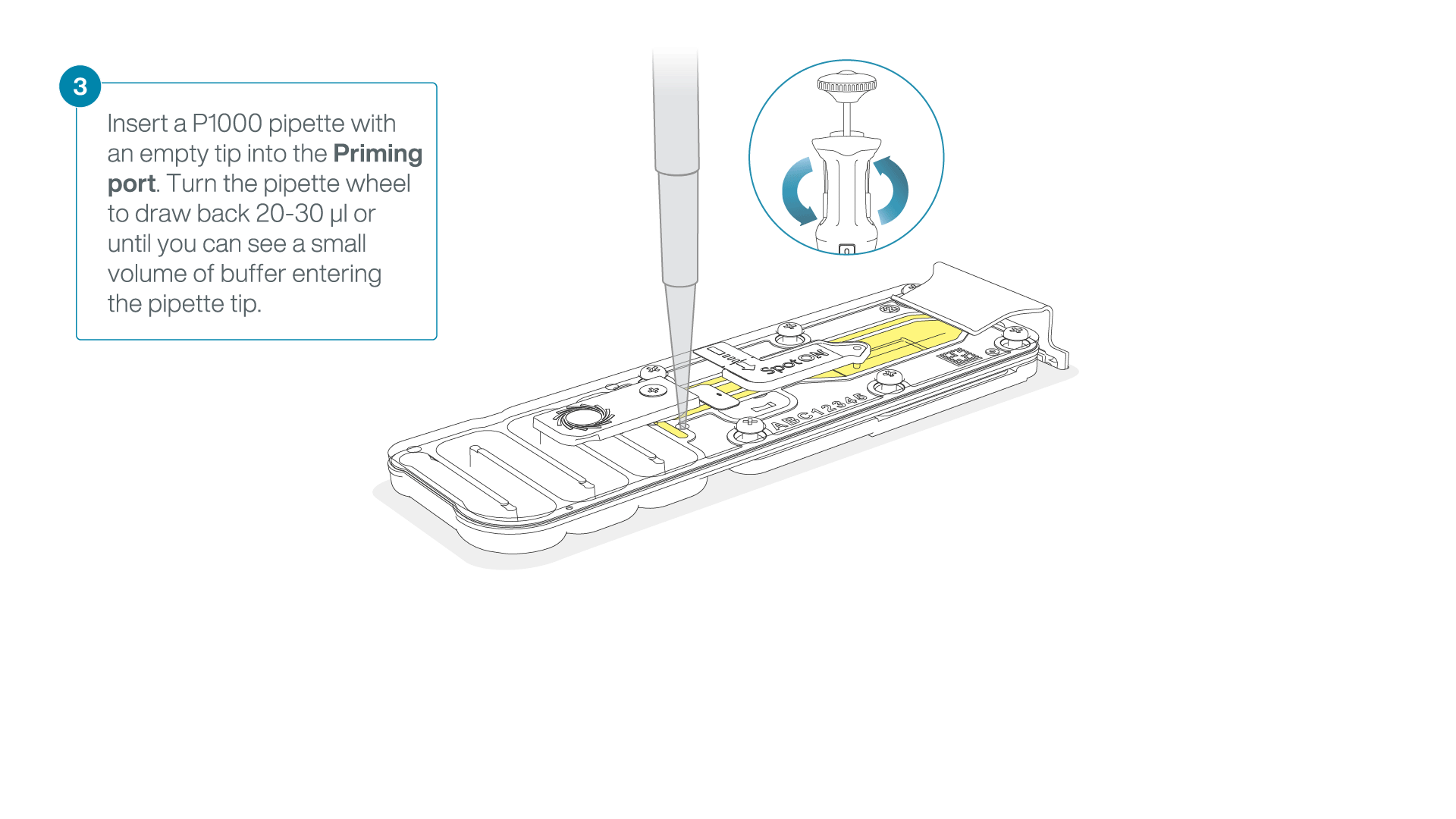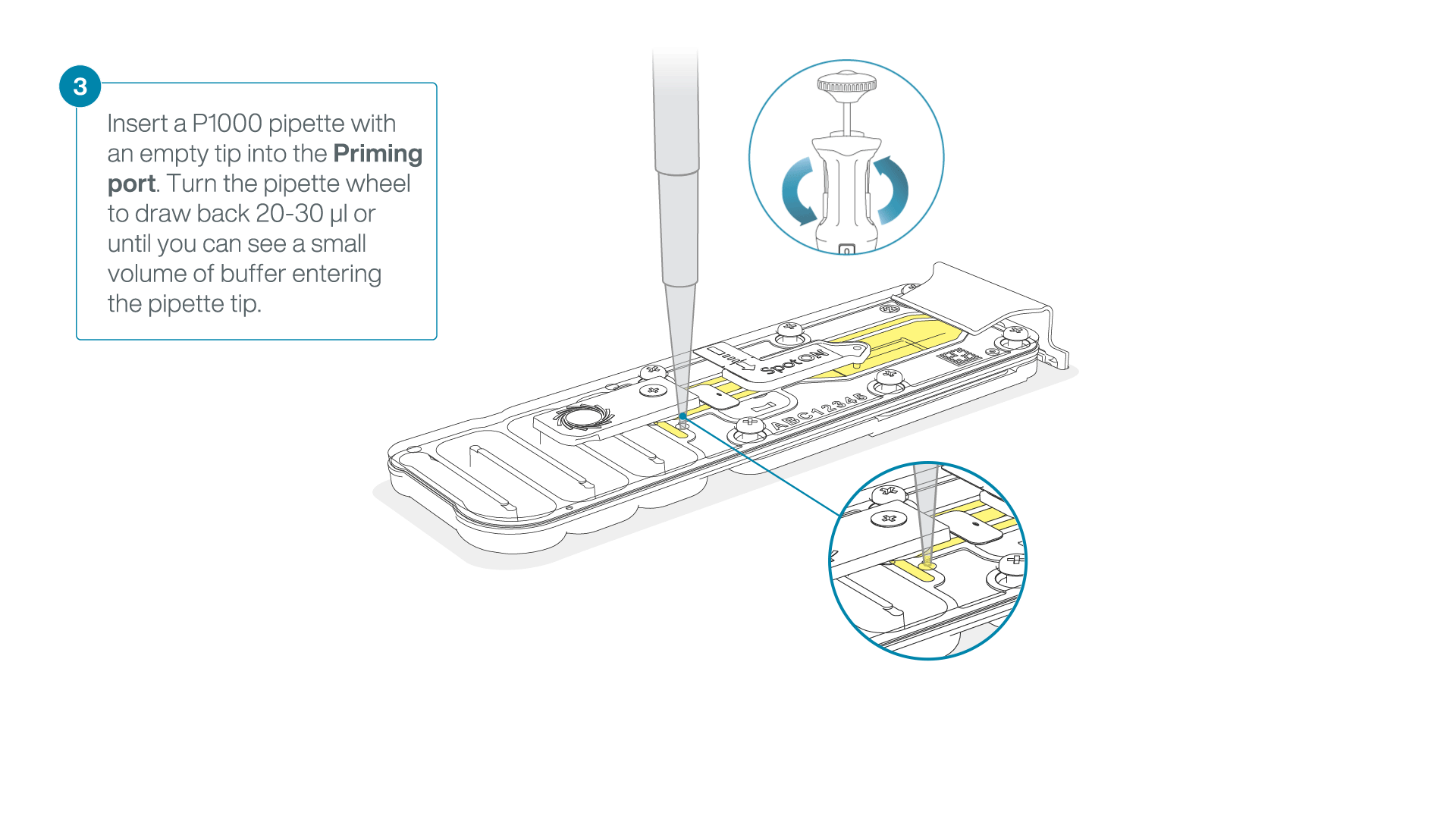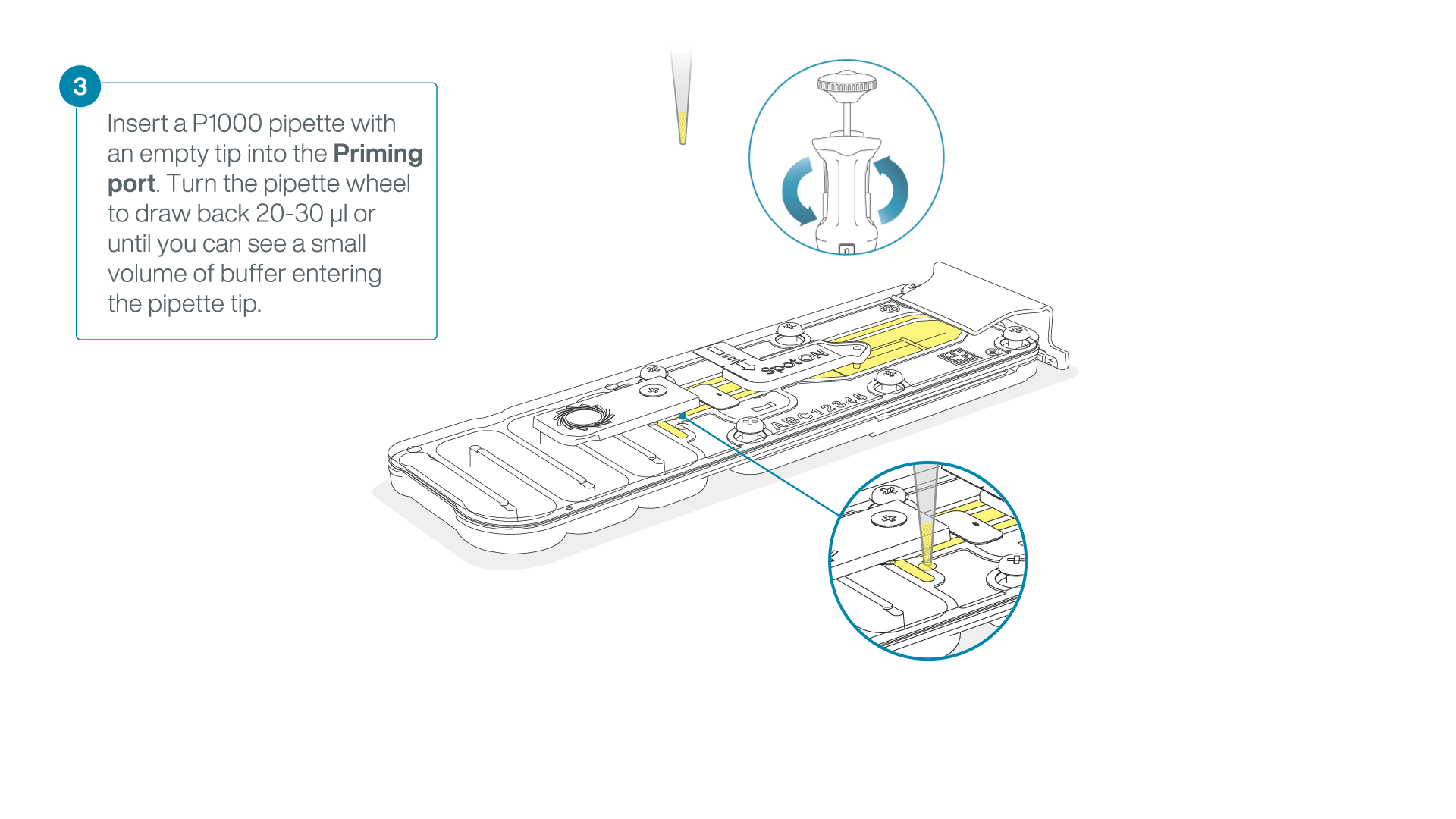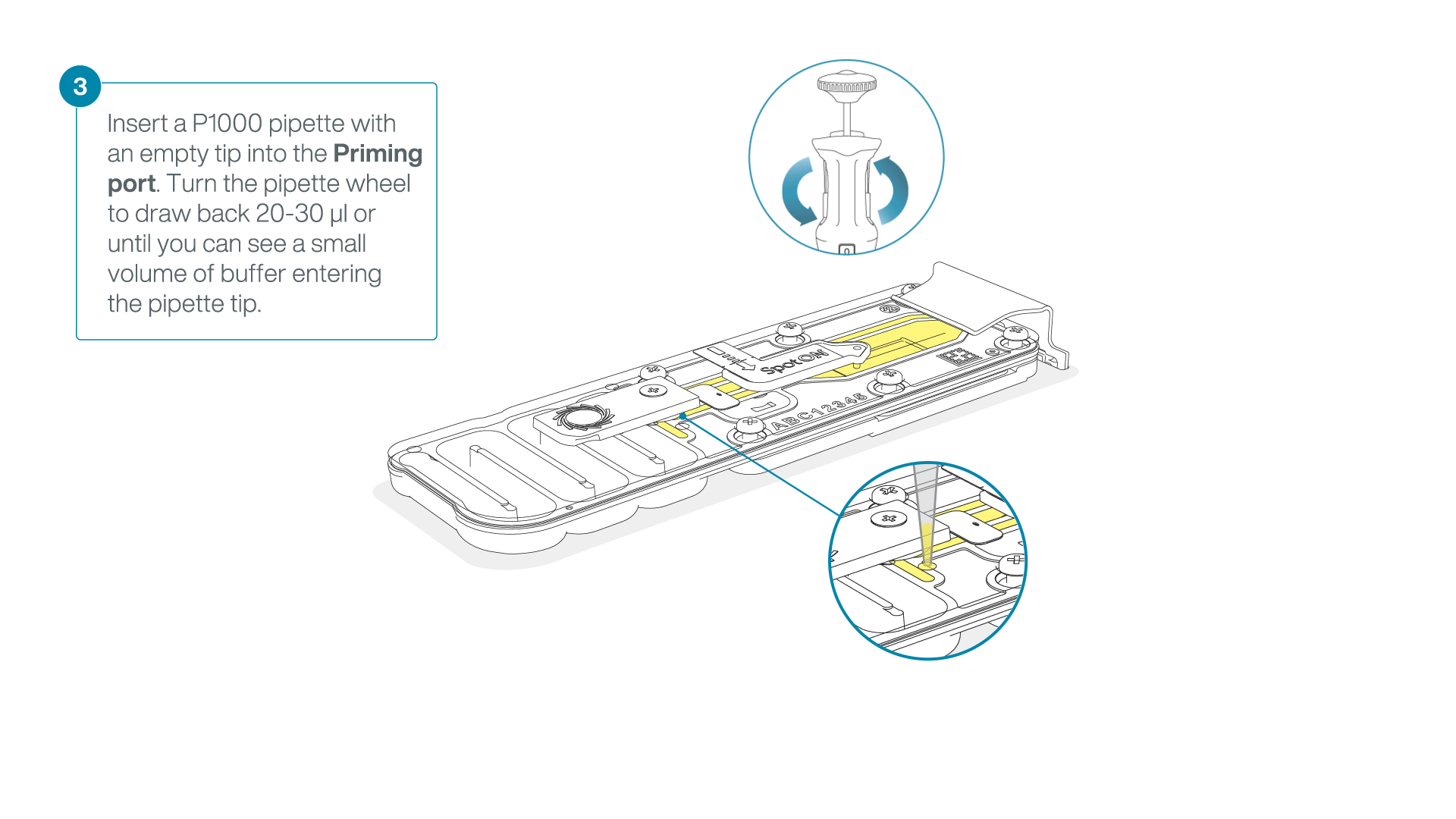
After opening the priming port, check for a small air bubble under the cover. Draw back a small volume to remove any bubbles:
- Set a P1000 pipette to 200 µl
- Insert the tip into the priming port
- Turn the wheel until the dial shows 220-230 µl, to draw back 20-30 µl, or until you can see a small volume of buffer entering the pipette tip
Note: Visually check that there is continuous buffer from the priming port across the sensor array.
-
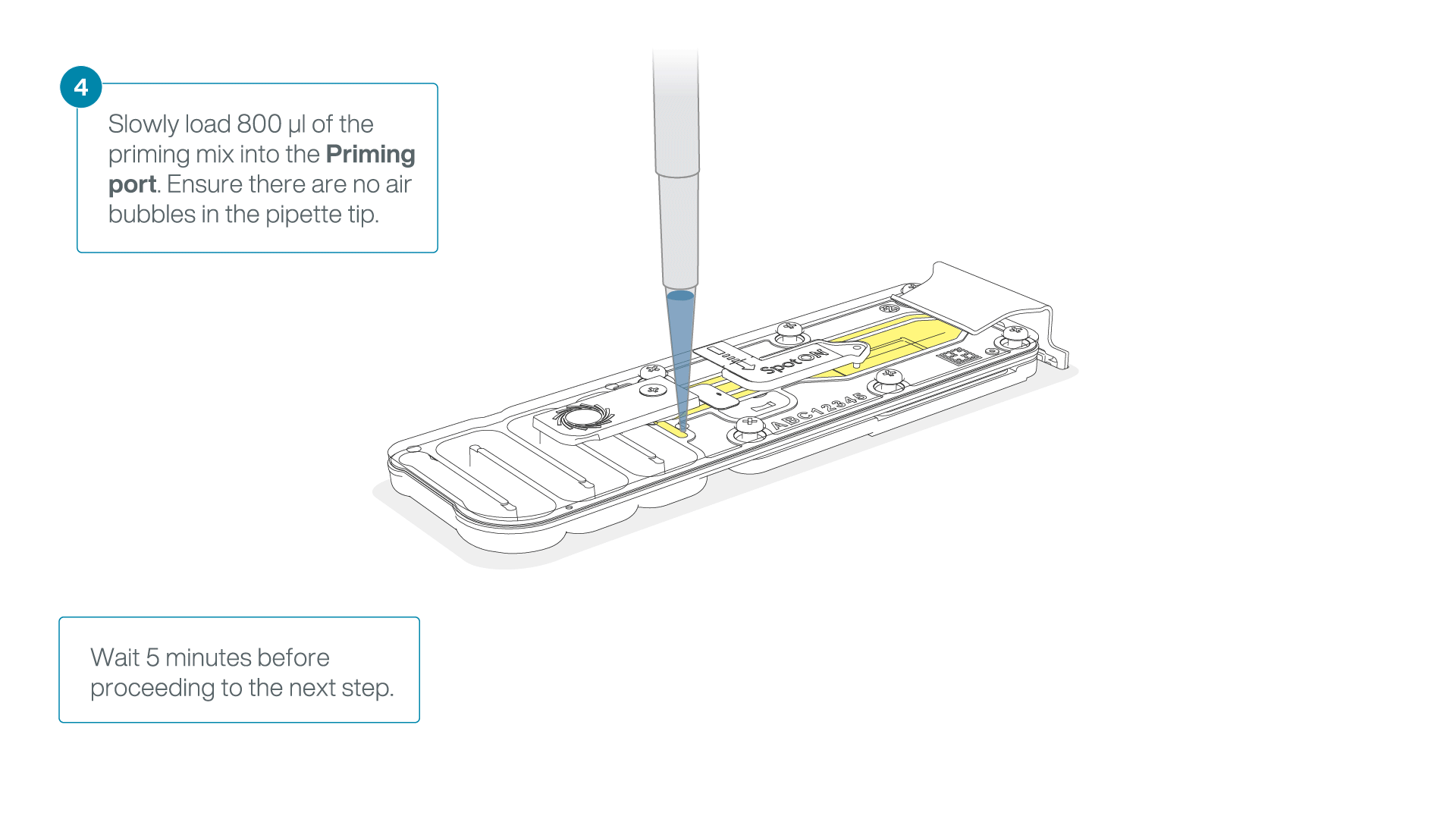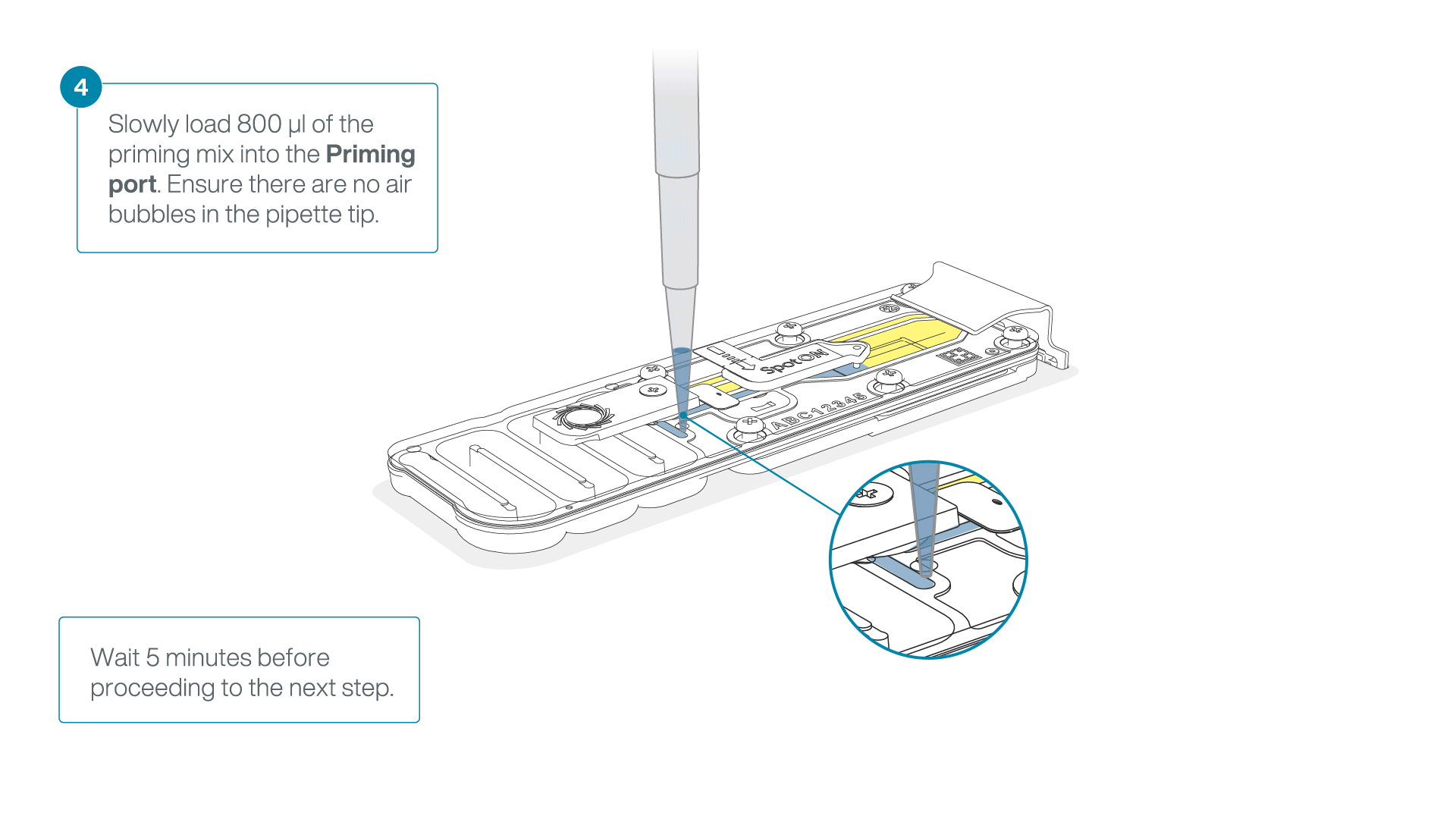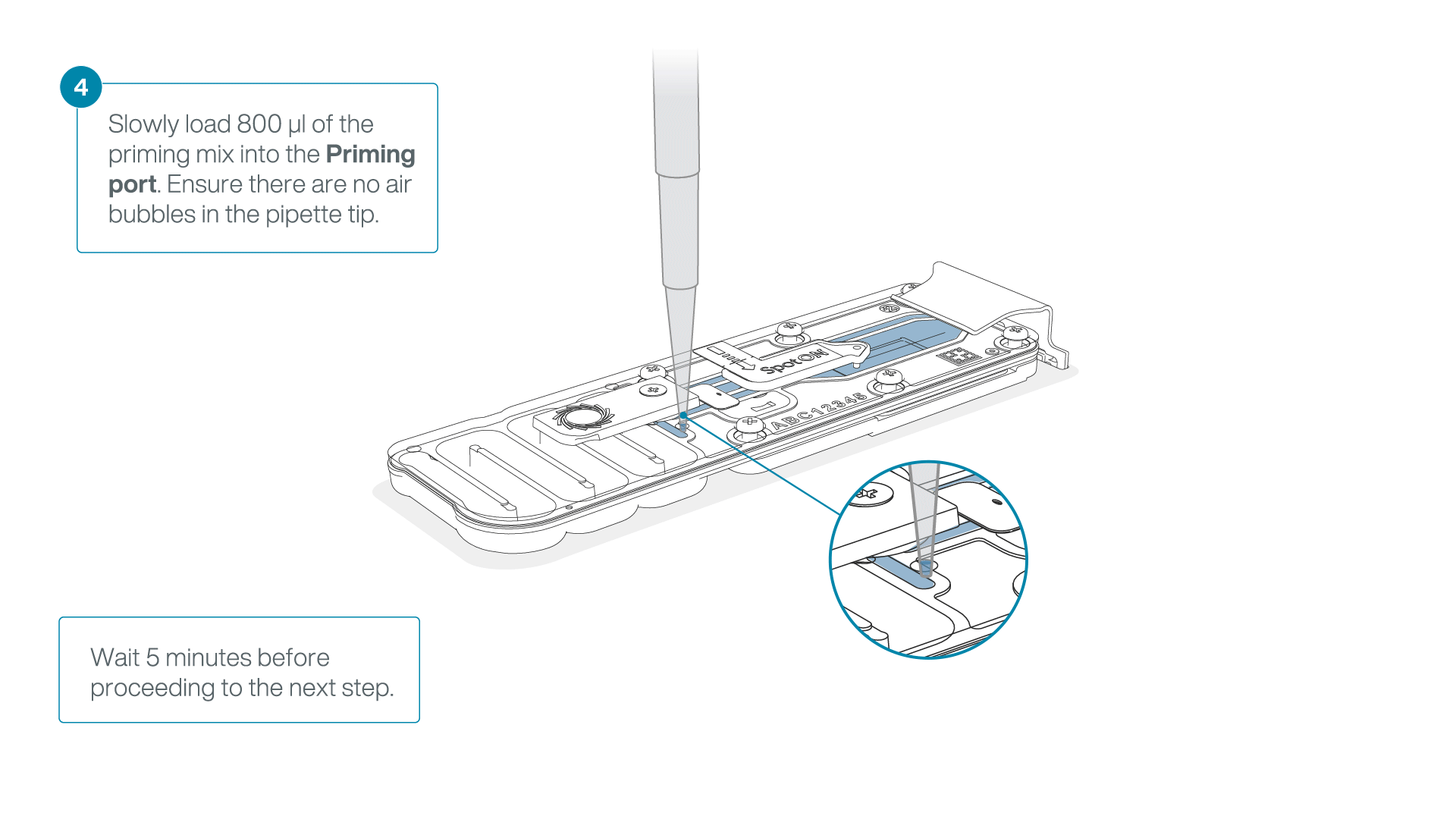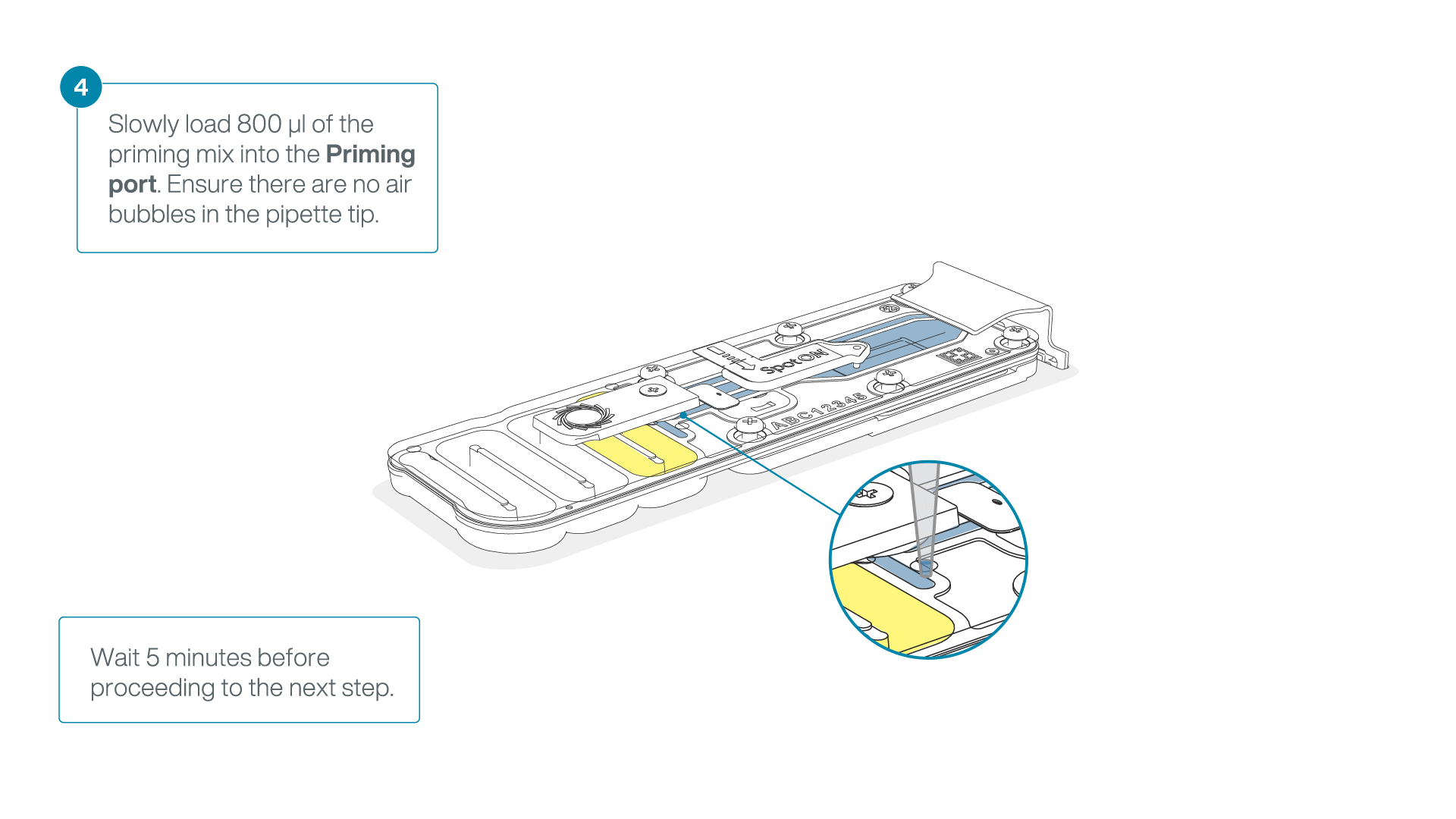
Load 800 µl of the priming mix into the flow cell via the priming port, avoiding the introduction of air bubbles. Wait for five minutes. During this time, prepare the library for loading by following the steps below.
-
Thoroughly mix the contents of the Loading Beads (LB) by pipetting.
-
In a new tube, prepare the library for loading as follows:
Reagent Volume per flow cell Sequencing Buffer (SQB) 37.5 µl Loading Beads (LB), mixed immediately before use 25.5 µl DNA library 12 µl Total 75 µl Note: Load the library onto the flow cell immediately after adding the Sequencing Buffer (SQB) and Loading Beads (LB) because the fuel in the buffer will start to be consumed by the adapter.
-
Complete the flow cell priming:
- Gently lift the SpotON sample port cover to make the SpotON sample port accessible.
- Load 200 µl of the priming mix into the flow cell priming port (not the SpotON sample port), avoiding the introduction of air bubbles.

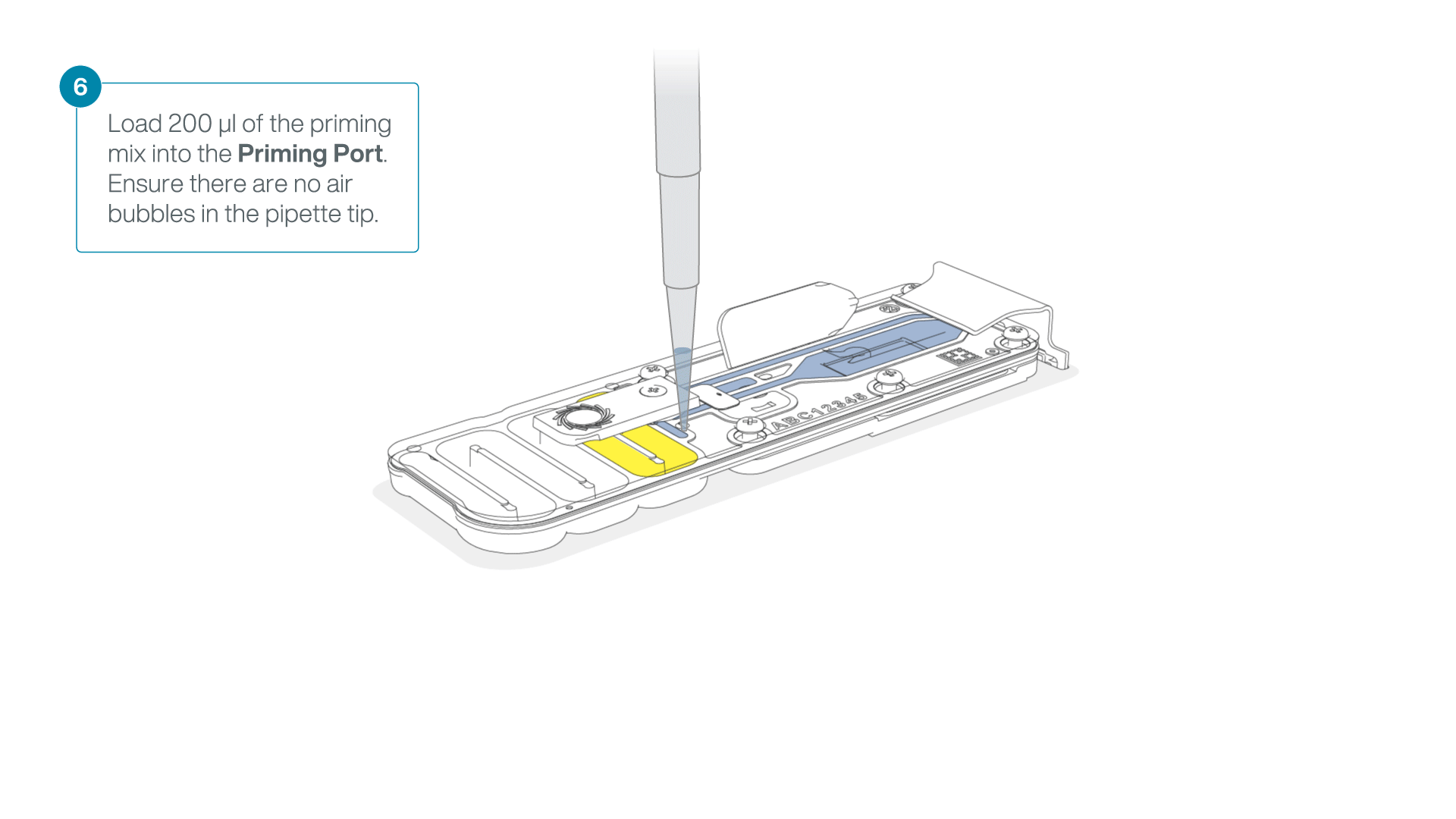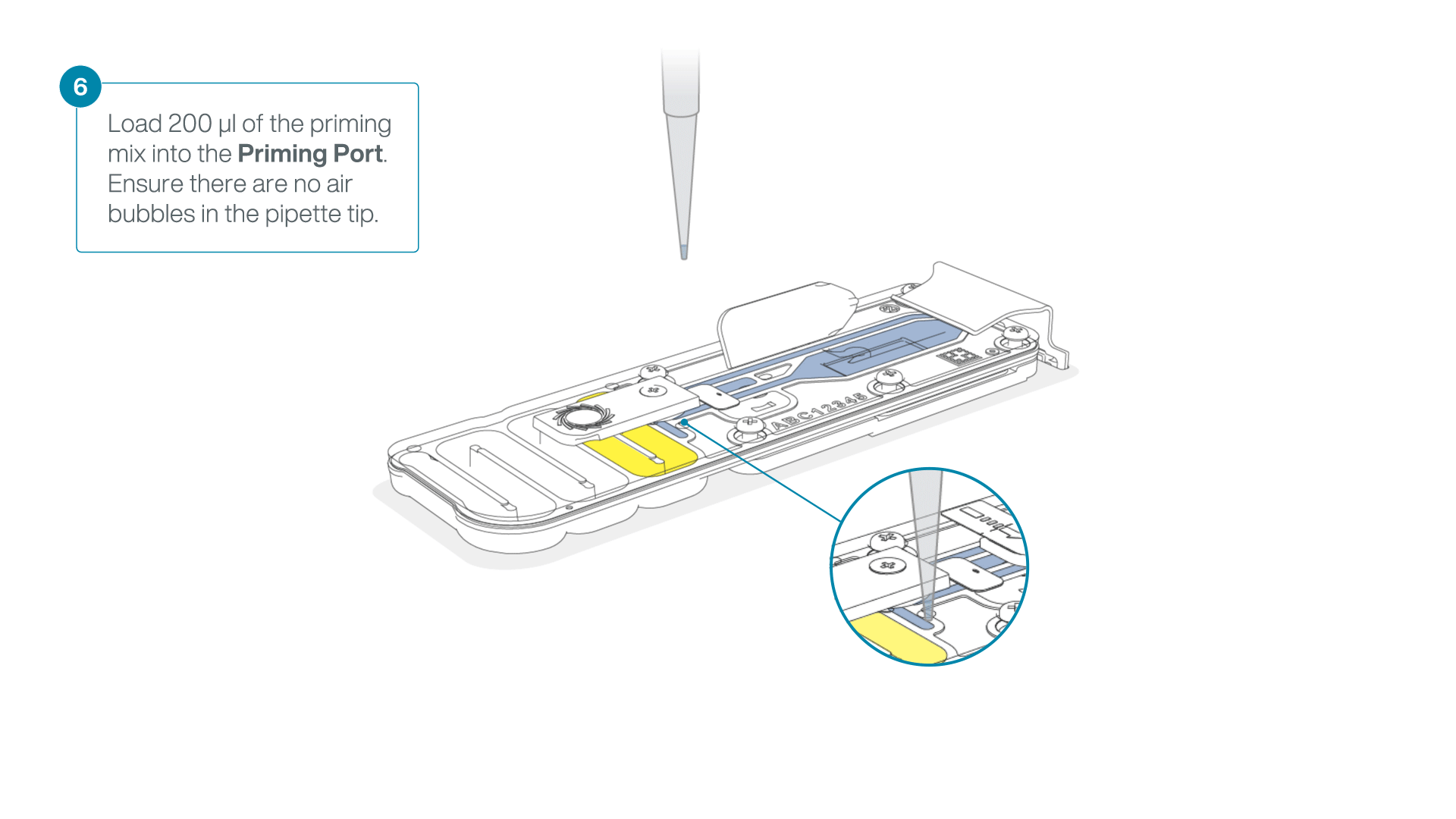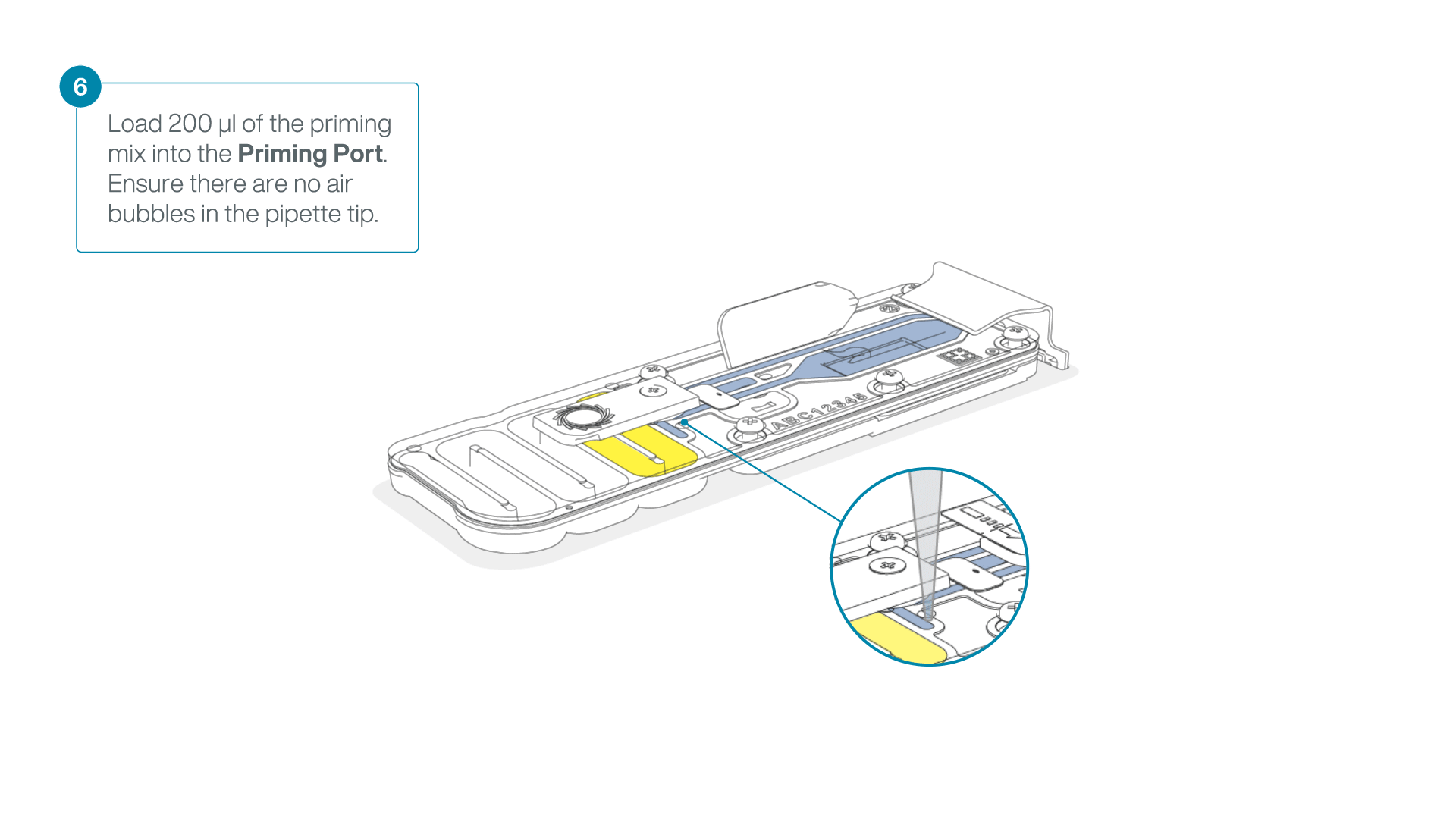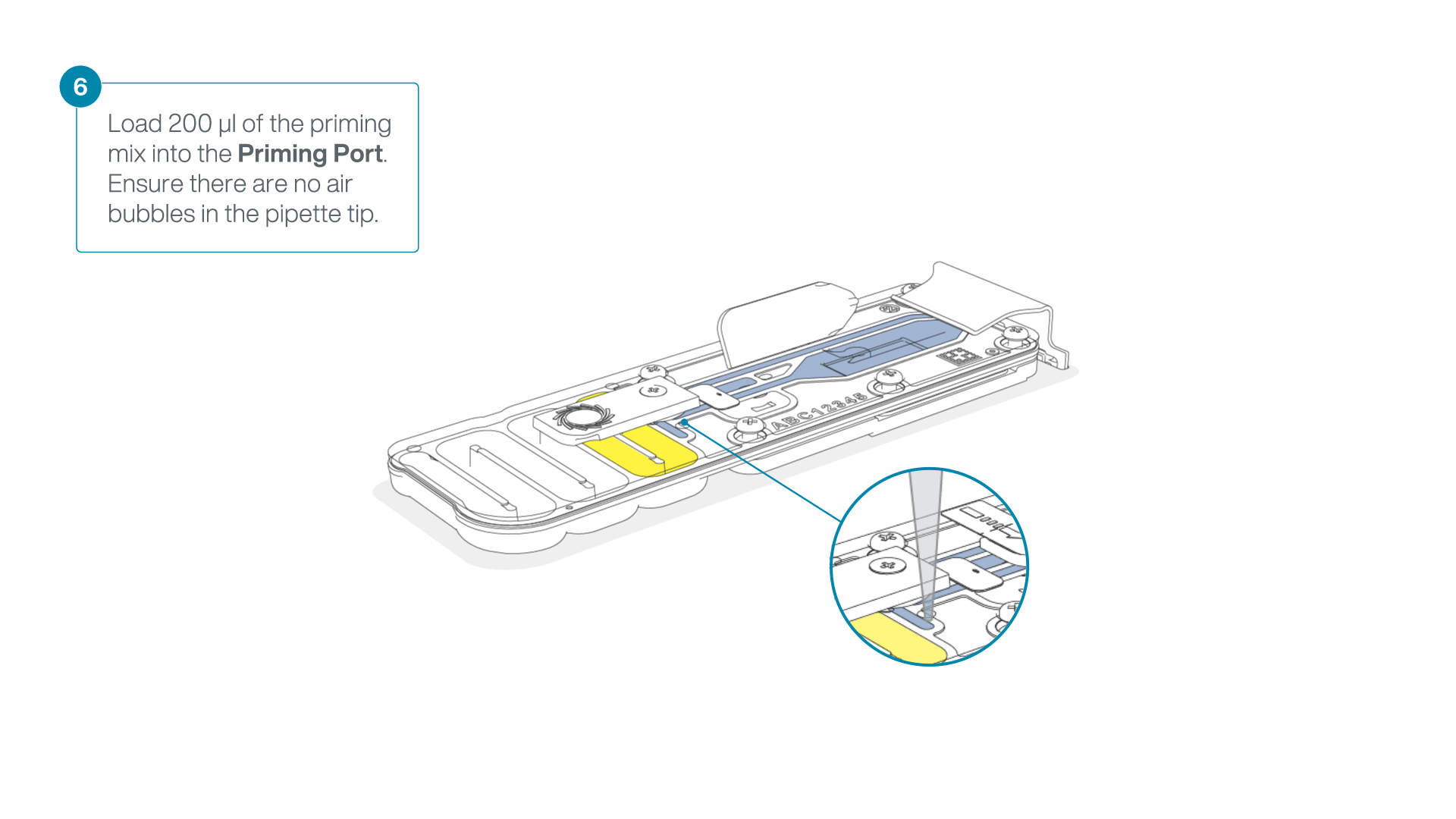
-
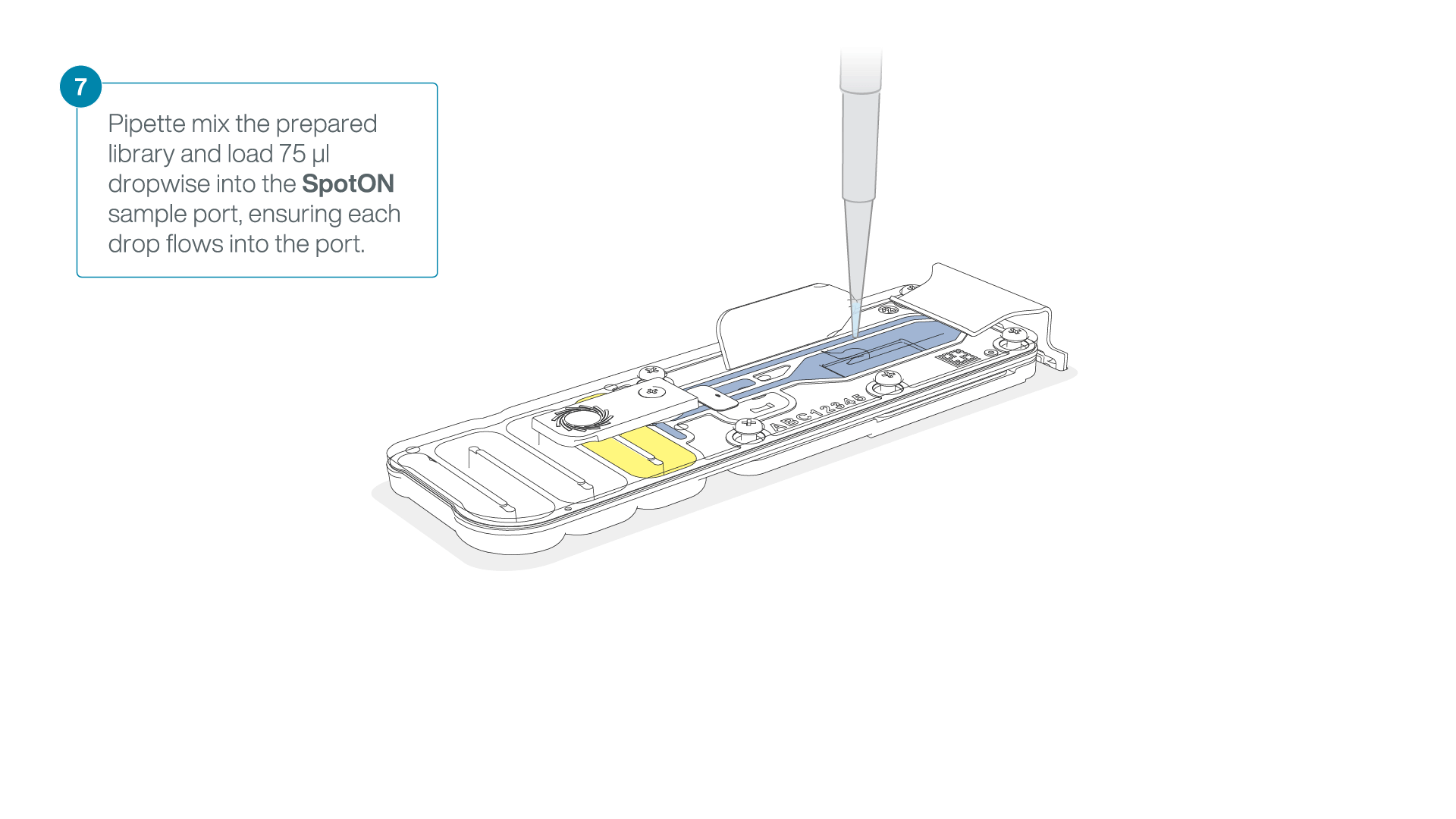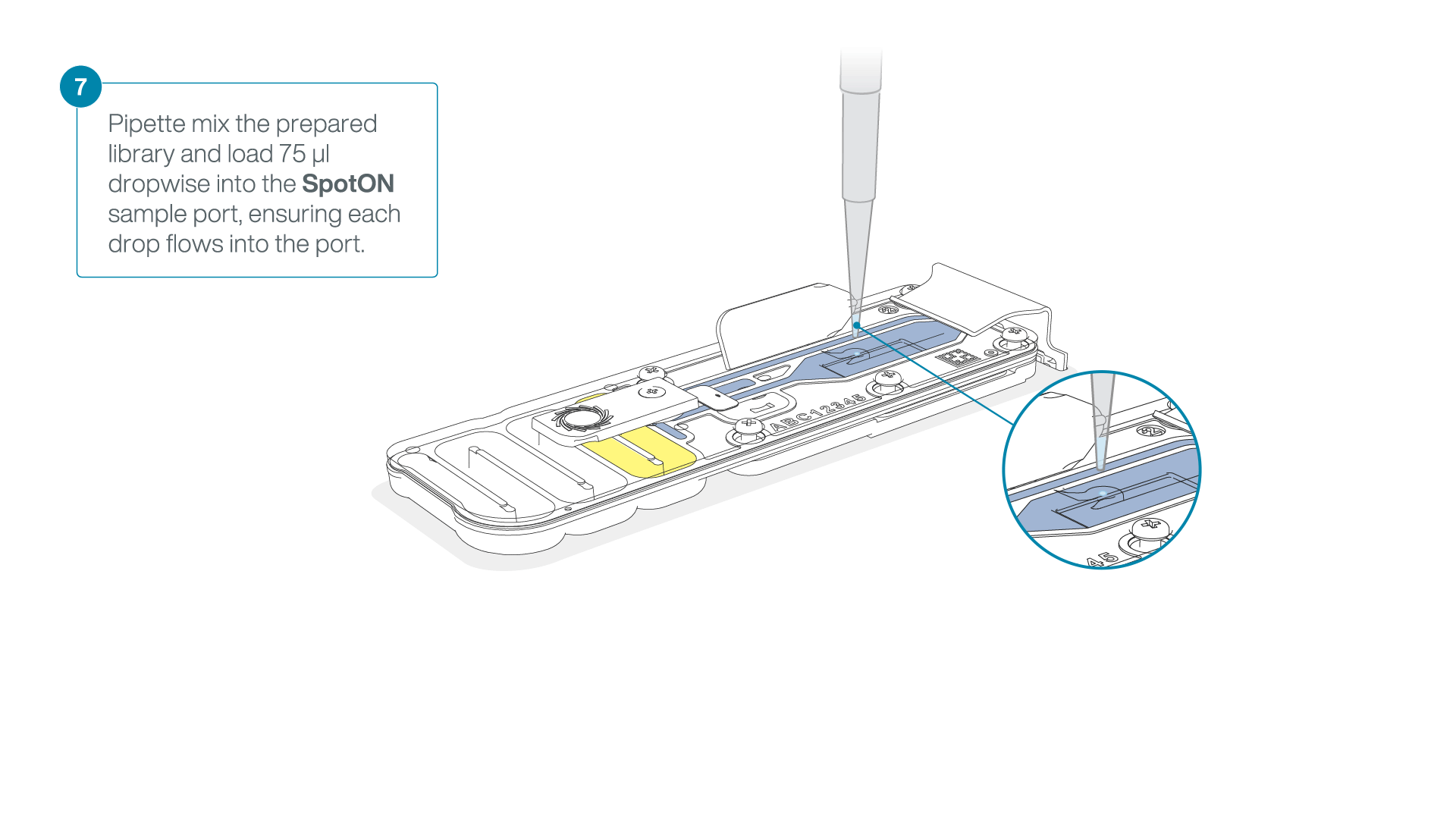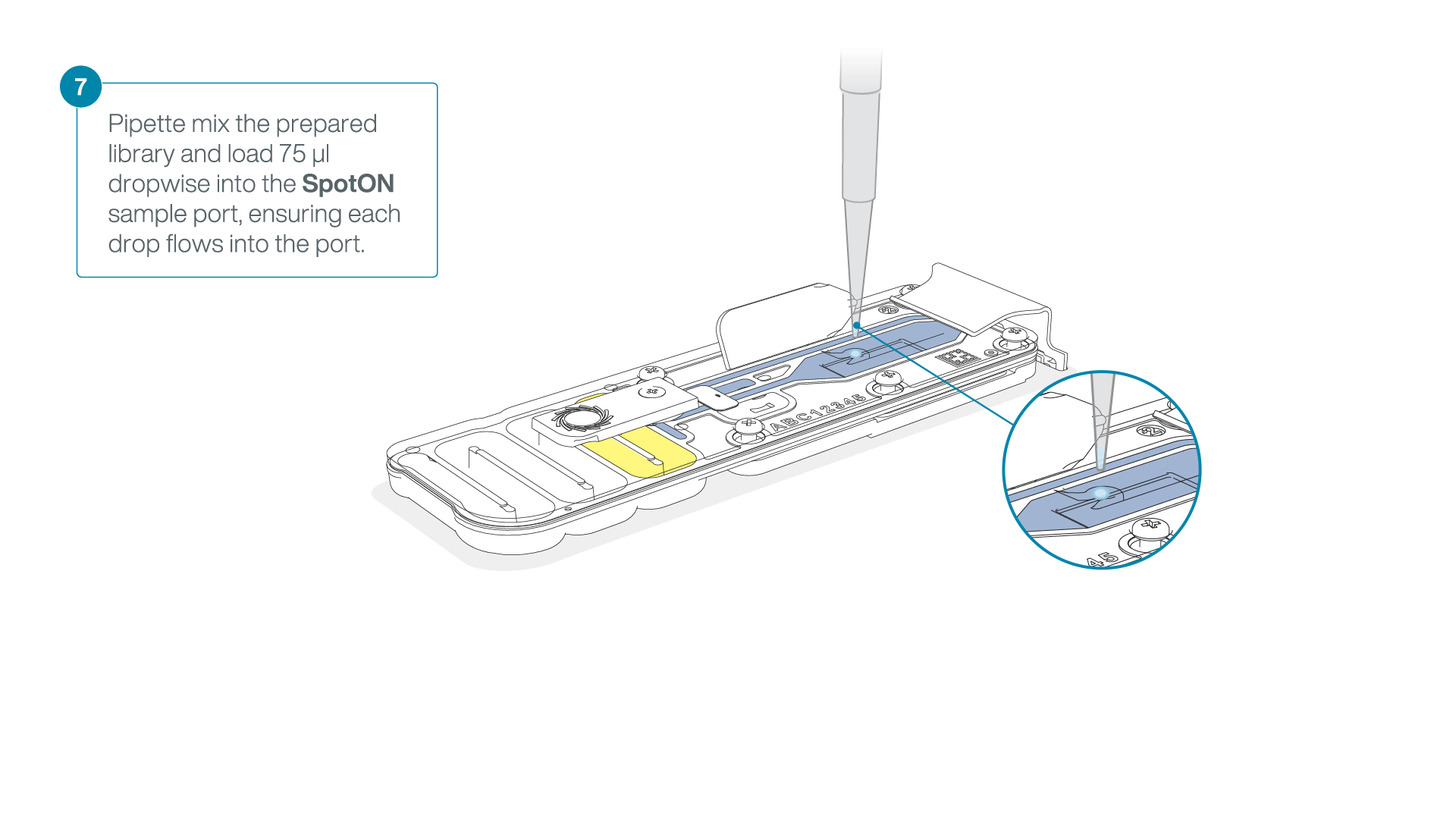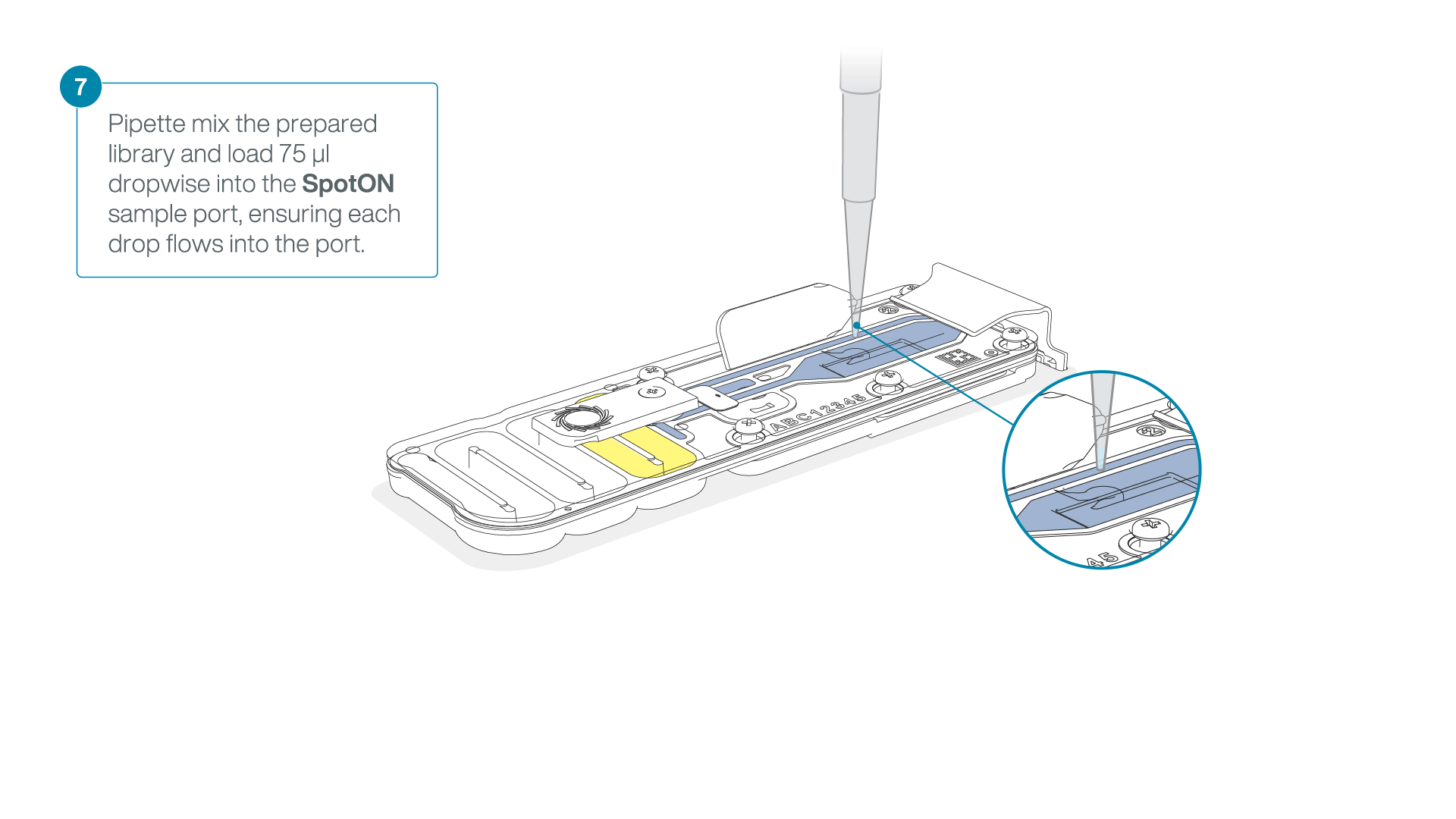
Mix the prepared library gently by pipetting up and down just prior to loading.
-
Add 75 μl of the prepared library to the flow cell via the SpotON sample port in a dropwise fashion. Ensure each drop flows into the port before adding the next.
-
Gently replace the SpotON sample port cover, making sure the bung enters the SpotON port and close the priming port.


-
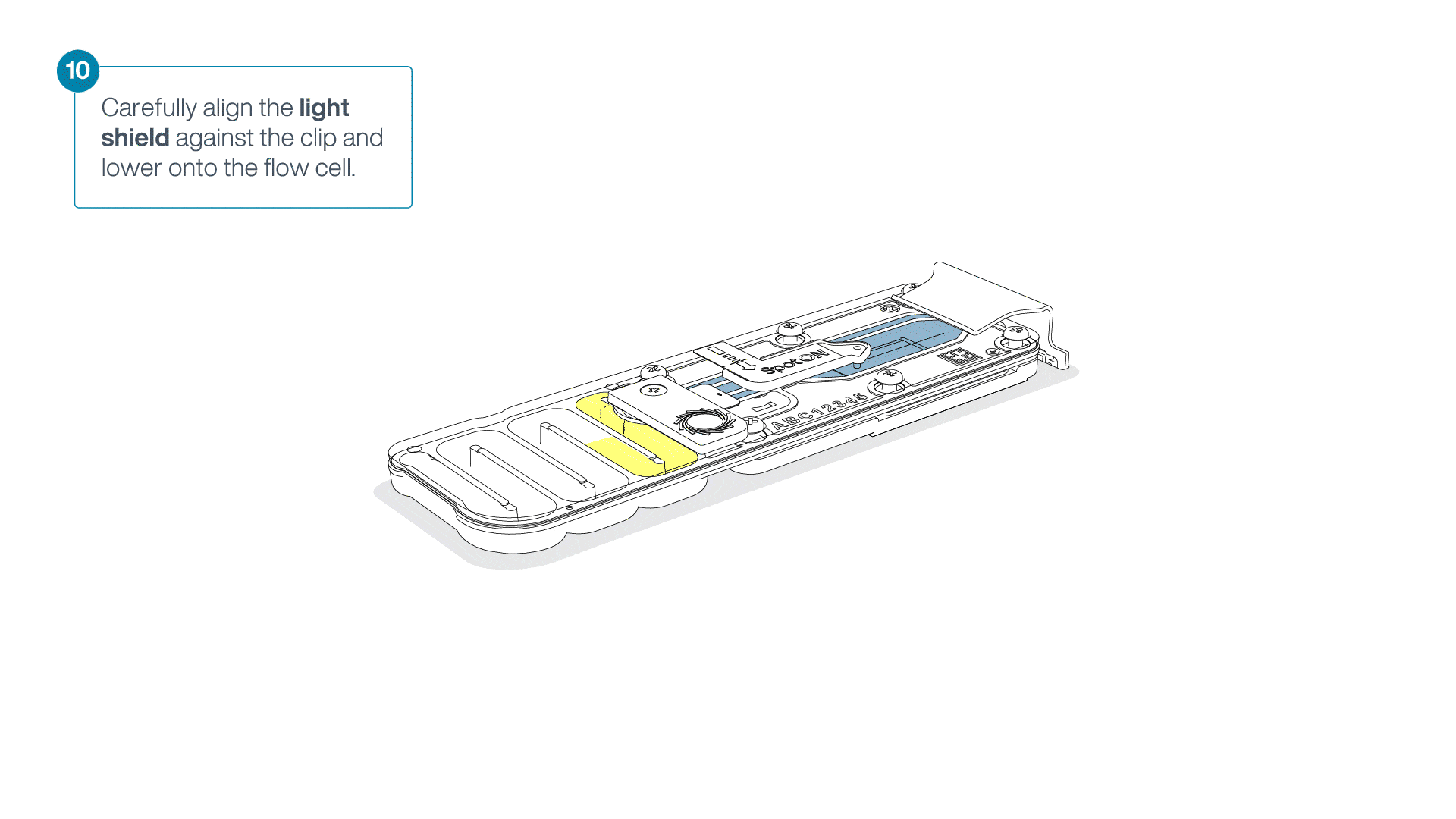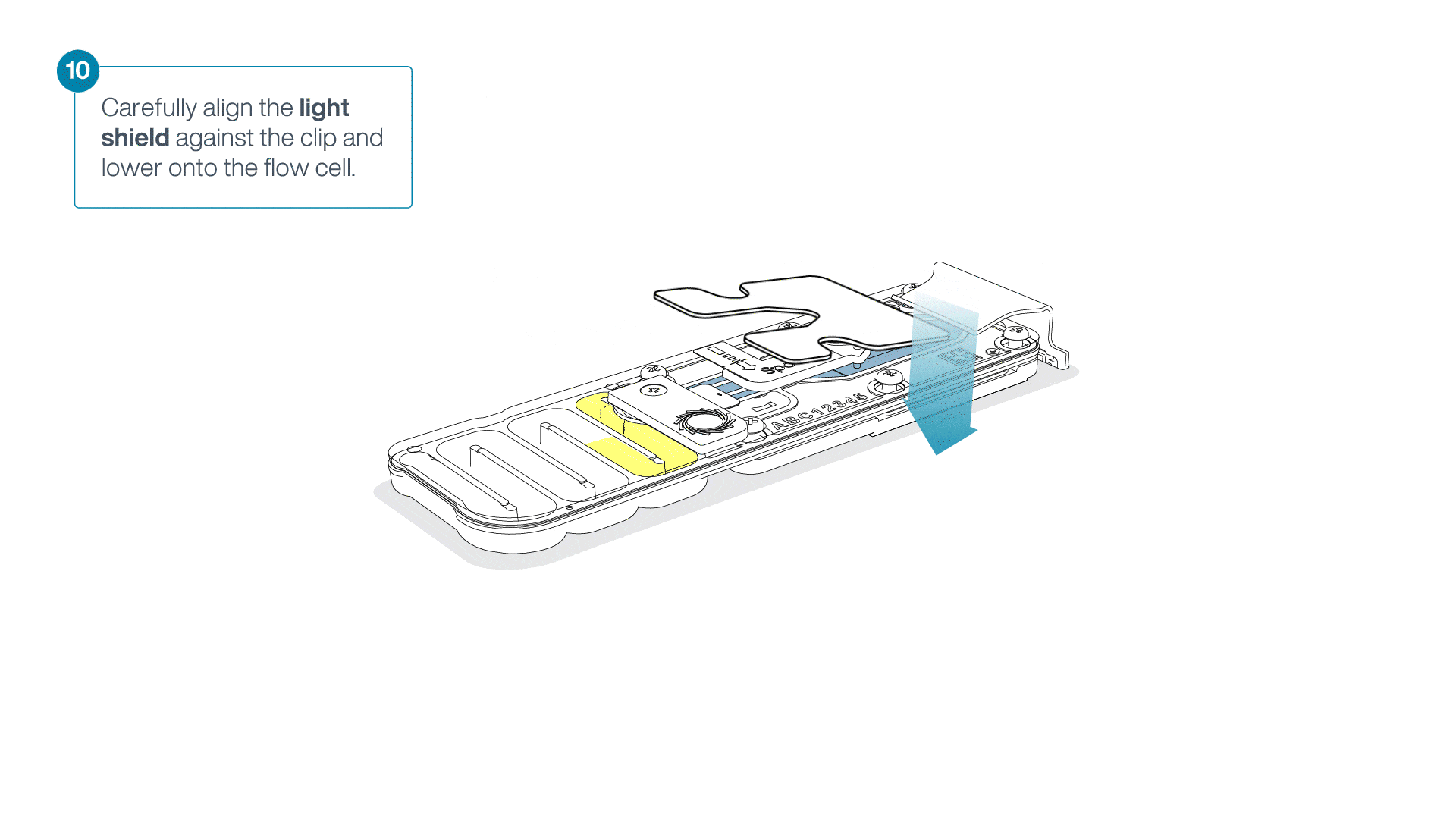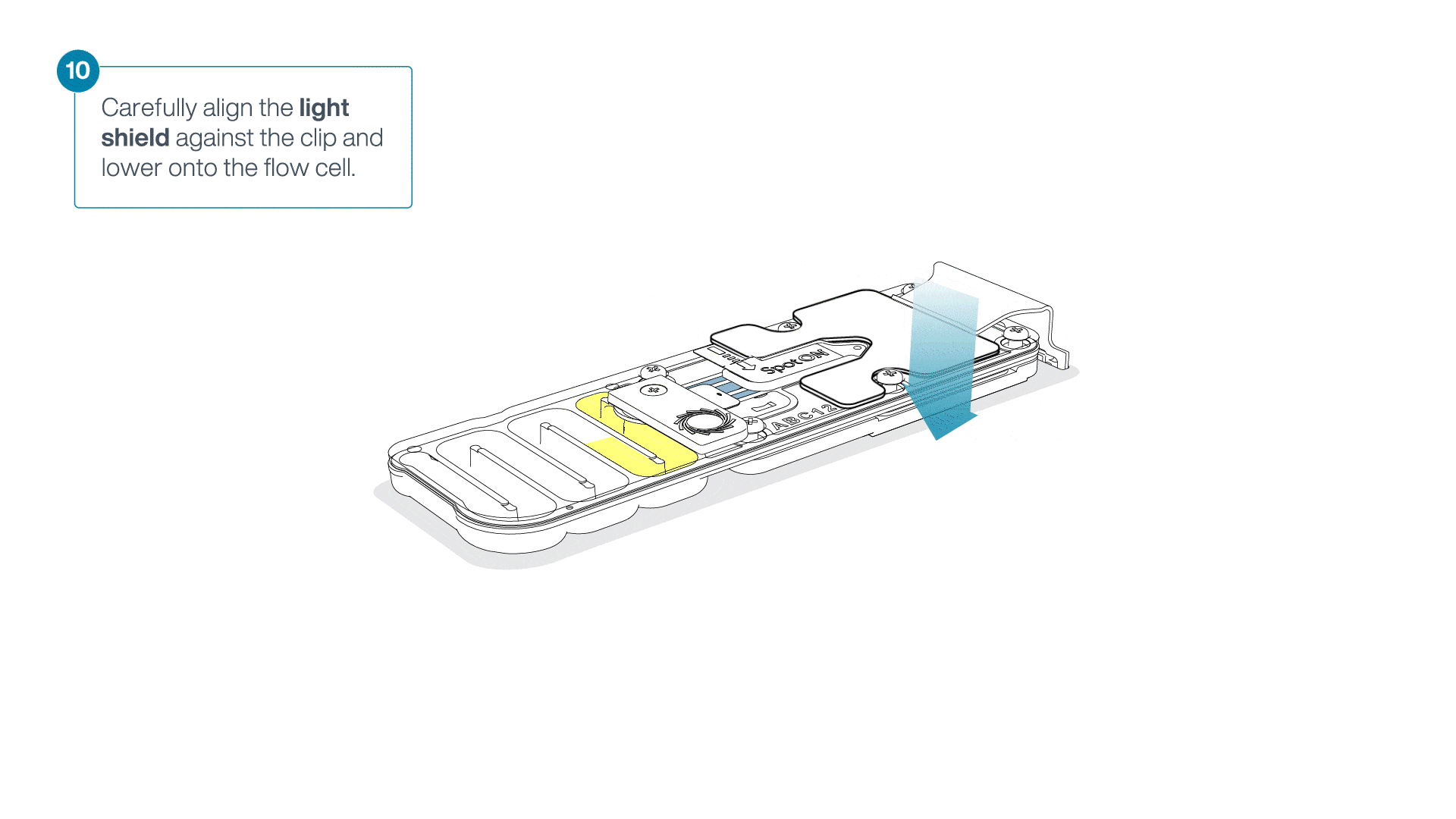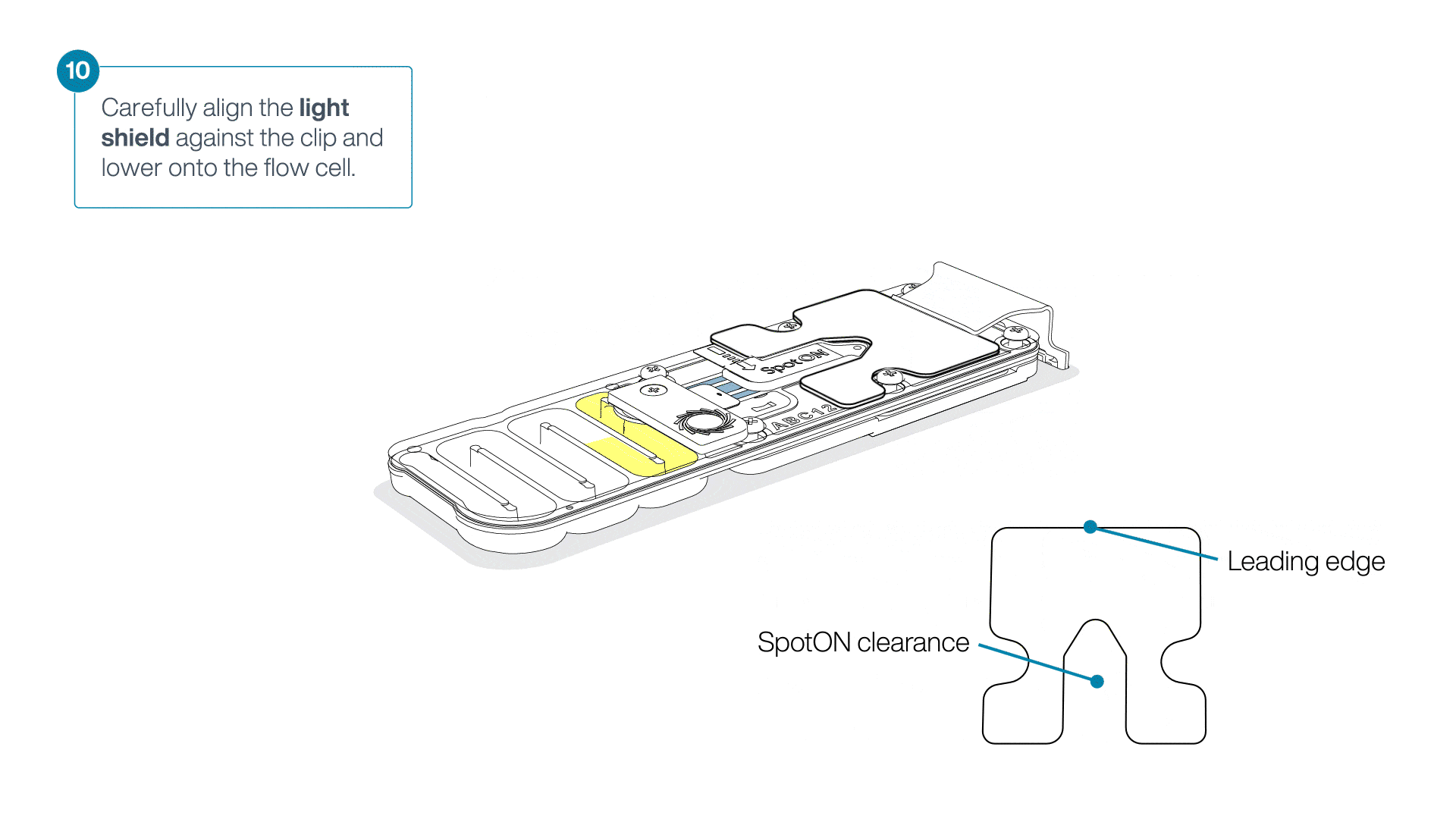
Place the light shield onto the flow cell, as follows:
Carefully place the leading edge of the light shield against the clip.
Note: Do not force the light shield underneath the clip.Gently lower the light shield onto the flow cell. The light shield should sit around the SpotON cover, covering the entire top section of the flow cell.











Sequencing and data analysis
Data acquisition and basecalling
Data acquisition and basecalling
-
Overview of nanopore data analysis
For a full overview of nanopore data analysis, which includes options for basecalling and post-basecalling analysis, please refer to the Data Analysis document.
-
How to start sequencing
The sequencing device control, data acquisition and real-time basecalling are carried out by the MinKNOW software. Please ensure MinKNOW is installed on your computer or device. There are multiple options for how to carry out sequencing:
1. Data acquisition and basecalling in real-time using MinKNOW on a computer
Follow the instructions in the MinKNOW protocol beginning from the "Starting a sequencing run" section until the end of the "Completing a MinKNOW run" section.
2. Data acquisition and basecalling in real-time using the MinION Mk1B/Mk1D device
Follow the instructions in the MinION Mk1B user manual or the MinION Mk1D user manual.
3. Data acquisition and basecalling in real-time using the MinION Mk1C device
Follow the instructions in the MinION Mk1C user manual.
4. Data acquisition and basecalling in real-time using the GridION device
Follow the instructions in the GridION user manual.
5. Data acquisition and basecalling in real-time using the PromethION device
Follow the instructions in the PromethION user manual or the PromethION 2 Solo user manual.
6. Data acquisition using MinKNOW on a computer and basecalling at a later time using MinKNOW
Follow the instructions in the MinKNOW protocol beginning from the "Starting a sequencing run" section until the end of the "Completing a MinKNOW run" section. When setting your experiment parameters, set the Basecalling tab to OFF. After the sequencing experiment has completed, follow the instructions in the Post-run analysis section of the MinKNOW protocol.
Downstream analysis
Downstream analysis
-
Additional resources for analysing your ASFV data
Note: Information subject to change, please refer to No part gets left behind: Tiled nanopore sequencing of whole ASFV genomes stitched together using Lilo by Amanda Warr et al., 2021 for most recent updates.
-
Bioinformatic processing of ASFV genomes sequenced with shotgun sequencing
The shotgun sequencing data were basecalled and demultiplexed using MinKNOW (v19.06.8) using Fast basecalling.
Following basecalling the reads were aligned to an ASFV genome using minimap2 to identify ASFV reads, the .fast5 files for these reads were extracted using fast5_subset from the ont_fast5_api and these again using high accuracy basecalling (this reduces basecalling time, suitable for when working with lower spec laptops/computers that have low GPU capacity).
The reads were assembled with Flye (v2.6) - additional Flye resources available following the link: Assembly of long, error-prone reads using repeat graphs and polished three times with Medaka (v0.7.1).
Comparisons of quantity of data produced and the proportion of which were ASFV reads were done using NanoComp (v1.28.1) - additional Nanopack resources available following the link: NanoPack: visualizing and processing long-read sequencing data. -
Bioinformatic processing of ASFV genomes from tiled amplicons with Lilo
The data were basecalled and demultiplexed using Guppy (v5.0.14) using high or super accuracy model on a GPU.
The snakemake pipeline Lilo was used, taking the following steps:
- Use Porechop (v0.2.3) to remove any sequencing adapters or barcodes that have made it through demultiplexing.
- Align to a reference with minimap2 (v2.22) and samtools (v1.12) and separate reads into amplicons by alignment position with bedtools (v2.30.0).
- Select reads of the expected amplicon length (+/-5%) and subset to 300X
- Select the read with highest average base quality within +/-1% of the median length of reads for the amplicon to be the “reference” using bioawk v1 and remove any amplicons with fewer than 40 reads (targeting the median length allows for flexibility for large insertions or deletions).
- Pool amplicon reads and references back into their original non-overlapping pools.
- Polish the pools three times with Medaka (v1.4.4) and combine resulting polished amplicons.
- Align to the reference with minimap2 and remove soft clipped bases (these likely represent missed barcodes or adapters).
- Run porechop to remove primers from the amplicons.
- Merge the amplicons with scaffold_builder (v2.3).
The required input to Lilo are demultiplexed reads in FASTQ format in a directory named “raw/”, a reference FASTA, a .bed file of primer alignments (as output by primal scheme), and a .csv of primer sequences (if there are ambiguous bases it is advised to expand them first) and a config file, described on the GitHub page. It is adaptable to any species (with a single genome fragment/chromosome) with any tiled primer scheme. The pipeline outputs a FASTA file containing the assembled genome.
-
ARTIC assemblies
A subset of genomes were also assembled using the ARTIC pipeline (v1.2.1) following the bioinformatics SOP using the Medaka method.
-
Quality control of assembled genomes
Quast (v5.0.2) was used to compare the assembled genomes to the most closely related publicly available ASFV assembly according to BLAST alignment (MN715134.1) - links and additional resources available following the link: Short and Long-Read Sequencing Survey of the Dynamic Transcriptomes of African Swine Fever Virus and the Host Cells.
Samples where both WGS and tiled sequencing were used were compared for overall structure using nucmer (v4.0.0beta2) - links and aditional reources available following the link: MUMmer4: A fast and versatile genome alignment system. -
Phylogeny
The phylogeny analysis was limited to the tiled genomes, as these were the most accurate assemblies, and publicly available genomes. These were aligned using Mafft (v7.467) and maximum likelihood trees constructed using iqtree (v2.0.5).
Flow cell reuse and returns
Flow cell reuse and returns
- Materials
-
- Flow Cell Wash Kit (EXP-WSH004)
-
After your sequencing experiment is complete, if you would like to reuse the flow cell, please follow the Flow Cell Wash Kit protocol and store the washed flow cell at +2°C to +8°C.
The Flow Cell Wash Kit protocol is available on the Nanopore Community.
-
Alternatively, follow the returns procedure to send the flow cell back to Oxford Nanopore.
Instructions for returning flow cells can be found here.
Troubleshooting
Issues during DNA extraction and library preparation
Issues during DNA extraction and library preparation
-
Below is a list of the most commonly encountered issues, with some suggested causes and solutions.
We also have an FAQ section available on the Nanopore Community Support section.
If you have tried our suggested solutions and the issue still persists, please contact Technical Support via email (support@nanoporetech.com) or via LiveChat in the Nanopore Community.
-
Low sample quality
Observation Possible cause Comments and actions Low DNA purity (Nanodrop reading for DNA OD 260/280 is <1.8 and OD 260/230 is <2.0–2.2) The DNA extraction method does not provide the required purity The effects of contaminants are shown in the Contaminants document. Please try an alternative extraction method that does not result in contaminant carryover.
Consider performing an additional SPRI clean-up step.Low RNA integrity (RNA integrity number <9.5 RIN, or the rRNA band is shown as a smear on the gel) The RNA degraded during extraction Try a different RNA extraction method. For more info on RIN, please see the RNA Integrity Number document. Further information can be found in the DNA/RNA Handling page. RNA has a shorter than expected fragment length The RNA degraded during extraction Try a different RNA extraction method. For more info on RIN, please see the RNA Integrity Number document. Further information can be found in the DNA/RNA Handling page.
We recommend working in an RNase-free environment, and to keep your lab equipment RNase-free when working with RNA. -
Low output from PCR
Observation Possible cause Comments and actions Low output from PCR Insufficient primer Increase the amount of primer pool in the PCR reaction by 0.25-1 µl, decrease water proportionally. - Insufficient DNA template Use undiluted DNA. - Thermocycler fault Ensure that you use a thermocycler that has been recently calibrated. - Insufficient viral DNA and an abundance of host DNA Use a host depletion method such as NEBNext® Microbiome DNA Enrichment Kit before PCR*. - Low representation of a particular primer in the sequencing data Spike in a small quantity of the primer in the 100 µM pool before future runs * This may be prohibitively expensive at scale, but for precious samples this will have a noticeable impact on amplicon yield.
-
Low DNA recovery after AMPure bead clean-up
Observation Possible cause Comments and actions Low recovery DNA loss due to a lower than intended AMPure beads-to-sample ratio 1. AMPure beads settle quickly, so ensure they are well resuspended before adding them to the sample.
2. When the AMPure beads-to-sample ratio is lower than 0.4:1, DNA fragments of any size will be lost during the clean-up.Low recovery DNA fragments are shorter than expected The lower the AMPure beads-to-sample ratio, the more stringent the selection against short fragments. Please always determine the input DNA length on an agarose gel (or other gel electrophoresis methods) and then calculate the appropriate amount of AMPure beads to use. 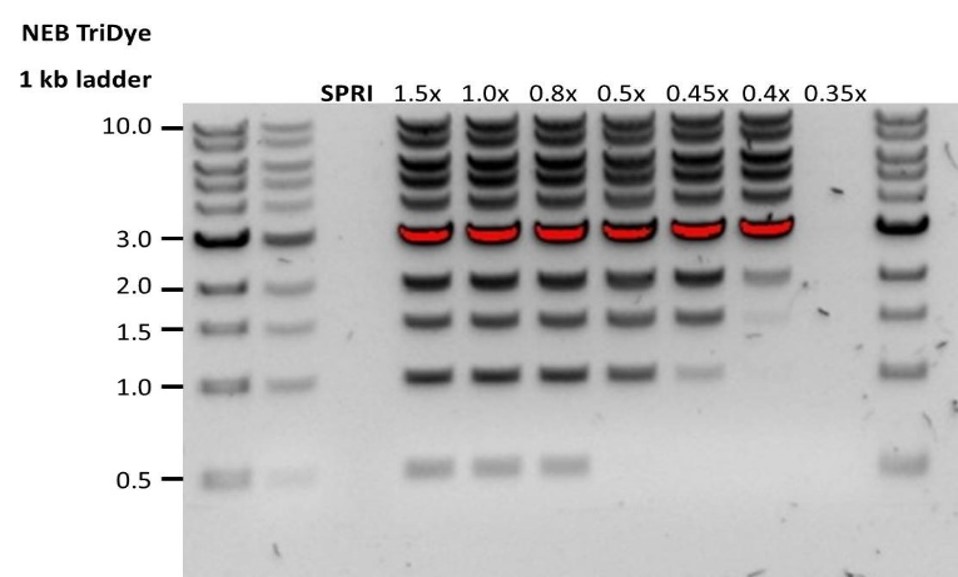
Low recovery after end-prep The wash step used ethanol <70% DNA will be eluted from the beads when using ethanol <70%. Make sure to use the correct percentage.

Issues during the sequencing run
Issues during the sequencing run
-
Below is a list of the most commonly encountered issues, with some suggested causes and solutions.
We also have an FAQ section available on the Nanopore Community Support section.
If you have tried our suggested solutions and the issue still persists, please contact Technical Support via email (support@nanoporetech.com) or via LiveChat in the Nanopore Community.
-
Fewer pores at the start of sequencing than after Flow Cell Check
Observation Possible cause Comments and actions MinKNOW reported a lower number of pores at the start of sequencing than the number reported by the Flow Cell Check An air bubble was introduced into the nanopore array After the Flow Cell Check it is essential to remove any air bubbles near the priming port before priming the flow cell. If not removed, the air bubble can travel to the nanopore array and irreversibly damage the nanopores that have been exposed to air. The best practice to prevent this from happening is demonstrated in this video. MinKNOW reported a lower number of pores at the start of sequencing than the number reported by the Flow Cell Check The flow cell is not correctly inserted into the device Stop the sequencing run, remove the flow cell from the sequencing device and insert it again, checking that the flow cell is firmly seated in the device and that it has reached the target temperature. If applicable, try a different position on the device (GridION/PromethION). MinKNOW reported a lower number of pores at the start of sequencing than the number reported by the Flow Cell Check Contaminations in the library damaged or blocked the pores The pore count during the Flow Cell Check is performed using the QC DNA molecules present in the flow cell storage buffer. At the start of sequencing, the library itself is used to estimate the number of active pores. Because of this, variability of about 10% in the number of pores is expected. A significantly lower pore count reported at the start of sequencing can be due to contaminants in the library that have damaged the membranes or blocked the pores. Alternative DNA/RNA extraction or purification methods may be needed to improve the purity of the input material. The effects of contaminants are shown in the Contaminants Know-how piece. Please try an alternative extraction method that does not result in contaminant carryover. -
MinKNOW script failed
Observation Possible cause Comments and actions MinKNOW shows "Script failed" Restart the computer and then restart MinKNOW. If the issue persists, please collect the MinKNOW log files and contact Technical Support. If you do not have another sequencing device available, we recommend storing the flow cell and the loaded library at 4°C and contact Technical Support for further storage guidance. -
Pore occupancy below 40%
Observation Possible cause Comments and actions Pore occupancy <40% Not enough library was loaded on the flow cell Ensure you load the recommended amount of good quality library in the relevant library prep protocol onto your flow cell. Please quantify the library before loading and calculate mols using tools like the Promega Biomath Calculator, choosing "dsDNA: µg to pmol" Pore occupancy close to 0 The Ligation Sequencing Kit was used, and sequencing adapters did not ligate to the DNA Make sure to use the NEBNext Quick Ligation Module (E6056) and Oxford Nanopore Technologies Ligation Buffer (LNB, provided in the sequencing kit) at the sequencing adapter ligation step, and use the correct amount of each reagent. A Lambda control library can be prepared to test the integrity of the third-party reagents. Pore occupancy close to 0 The Ligation Sequencing Kit was used, and ethanol was used instead of LFB or SFB at the wash step after sequencing adapter ligation Ethanol can denature the motor protein on the sequencing adapters. Make sure the LFB or SFB buffer was used after ligation of sequencing adapters. Pore occupancy close to 0 No tether on the flow cell Tethers are adding during flow cell priming (FLT/FCT tube). Make sure FLT/FCT was added to FB/FCF before priming. -
Shorter than expected read length
Observation Possible cause Comments and actions Shorter than expected read length Unwanted fragmentation of DNA sample Read length reflects input DNA fragment length. Input DNA can be fragmented during extraction and library prep.
1. Please review the Extraction Methods in the Nanopore Community for best practice for extraction.
2. Visualise the input DNA fragment length distribution on an agarose gel before proceeding to the library prep.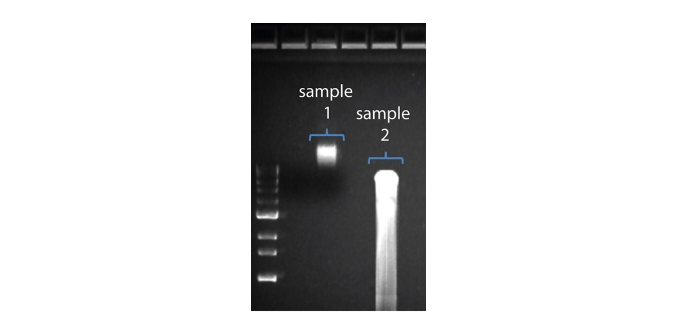 In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented.
In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented.
3. During library prep, avoid pipetting and vortexing when mixing reagents. Flicking or inverting the tube is sufficient. -
Large proportion of unavailable pores
Observation Possible cause Comments and actions Large proportion of unavailable pores (shown as blue in the channels panel and pore activity plot) 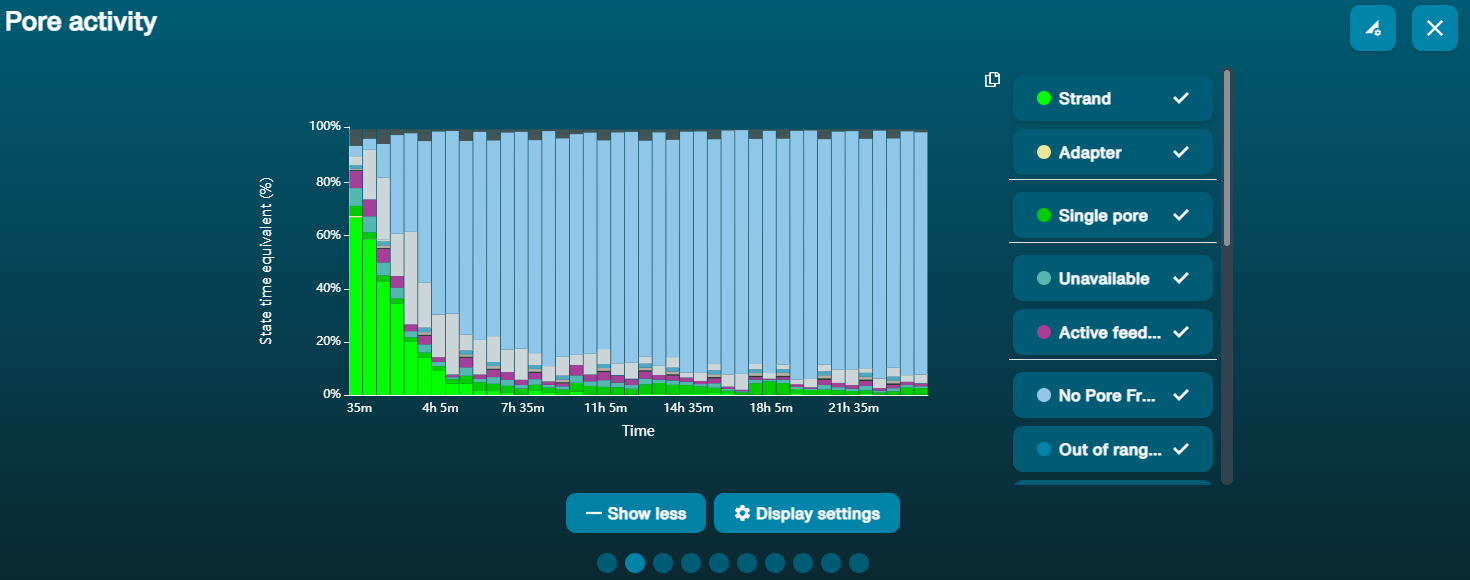 The pore activity plot above shows an increasing proportion of "unavailable" pores over time.
The pore activity plot above shows an increasing proportion of "unavailable" pores over time.Contaminants are present in the sample Some contaminants can be cleared from the pores by the unblocking function built into MinKNOW. If this is successful, the pore status will change to "sequencing pore". If the portion of unavailable pores stays large or increases:
1. A nuclease flush using the Flow Cell Wash Kit (EXP-WSH004) can be performed, or
2. Run several cycles of PCR to try and dilute any contaminants that may be causing problems. -
Large proportion of inactive pores
Observation Possible cause Comments and actions Large proportion of inactive/unavailable pores (shown as light blue in the channels panel and pore activity plot. Pores or membranes are irreversibly damaged) Air bubbles have been introduced into the flow cell Air bubbles introduced through flow cell priming and library loading can irreversibly damage the pores. Watch the Priming and loading your flow cell video for best practice Large proportion of inactive/unavailable pores Certain compounds co-purified with DNA Known compounds, include polysaccharides, typically associate with plant genomic DNA.
1. Please refer to the Plant leaf DNA extraction method.
2. Clean-up using the QIAGEN PowerClean Pro kit.
3. Perform a whole genome amplification with the original gDNA sample using the QIAGEN REPLI-g kit.Large proportion of inactive/unavailable pores Contaminants are present in the sample The effects of contaminants are shown in the Contaminants Know-how piece. Please try an alternative extraction method that does not result in contaminant carryover. -
Reduction in sequencing speed and q-score later into the run
Observation Possible cause Comments and actions Reduction in sequencing speed and q-score later into the run For Kit 9 chemistry (e.g. SQK-LSK109), fast fuel consumption is typically seen when the flow cell is overloaded with library (please see the appropriate protocol for your DNA library to see the recommendation). Add more fuel to the flow cell by following the instructions in the MinKNOW protocol. In future experiments, load lower amounts of library to the flow cell. -
Temperature fluctuation
Observation Possible cause Comments and actions Temperature fluctuation The flow cell has lost contact with the device Check that there is a heat pad covering the metal plate on the back of the flow cell. Re-insert the flow cell and press it down to make sure the connector pins are firmly in contact with the device. If the problem persists, please contact Technical Services. -
Failed to reach target temperature
Observation Possible cause Comments and actions MinKNOW shows "Failed to reach target temperature" The instrument was placed in a location that is colder than normal room temperature, or a location with poor ventilation (which leads to the flow cells overheating) MinKNOW has a default timeframe for the flow cell to reach the target temperature. Once the timeframe is exceeded, an error message will appear and the sequencing experiment will continue. However, sequencing at an incorrect temperature may lead to a decrease in throughput and lower q-scores. Please adjust the location of the sequencing device to ensure that it is placed at room temperature with good ventilation, then re-start the process in MinKNOW. Please refer to this link for more information on MinION temperature control. -
Guppy – no input .fast5 was found or basecalled
Observation Possible cause Comments and actions No input .fast5 was found or basecalled input_path did not point to the .fast5 file location The --input_path has to be followed by the full file path to the .fast5 files to be basecalled, and the location has to be accessible either locally or remotely through SSH. No input .fast5 was found or basecalled The .fast5 files were in a subfolder at the input_path location To allow Guppy to look into subfolders, add the --recursive flag to the command -
Guppy – no Pass or Fail folders were generated after basecalling
Observation Possible cause Comments and actions No Pass or Fail folders were generated after basecalling The --qscore_filtering flag was not included in the command The --qscore_filtering flag enables filtering of reads into Pass and Fail folders inside the output folder, based on their strand q-score. When performing live basecalling in MinKNOW, a q-score of 7 (corresponding to a basecall accuracy of ~80%) is used to separate reads into Pass and Fail folders. -
Guppy – unusually slow processing on a GPU computer
Observation Possible cause Comments and actions Unusually slow processing on a GPU computer The --device flag wasn't included in the command The --device flag specifies a GPU device to use for accelerate basecalling. If not included in the command, GPU will not be used. GPUs are counted from zero. An example is --device cuda:0 cuda:1, when 2 GPUs are specified to use by the Guppy command.
 In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented.
In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented. The pore activity plot above shows an increasing proportion of "unavailable" pores over time.
The pore activity plot above shows an increasing proportion of "unavailable" pores over time.Become a full member
Purchase a MinION Starter Pack from Avantor to get full community access and benefit from:
- News - hear about the latest product updates
- Posts - interact with thousands of nanopore users from around the globe
- Software - download the latest sequencing and analysis software
Already have a Nanopore Community account?
Log in hereNeed more help?
Request a call with our experts for detailed advice on implementing nanopore sequencing.
Request a callInterested in microbiology?
Visit our microbial sequencing spotlight page on vwr.com.
Microbial sequencing