Welcome to the Nanopore Community
Order MinION devices and consumables
Visit vwr.comLigation sequencing DNA XL V14 (SQK-LSK114-XL)
Version for device: MinION
Introduction to the protocol
Overview of the protocol
Overview of the protocol
-
Introduction to the Ligation Sequencing Kit XL V14 (SQK-LSK114-XL) protocol
This protocol describes how to carry out sequencing of multiple DNA samples simultaneously using the Ligation Sequencing Kit XL V14 (SQK-LSK114-XL). It is recommended that a Lambda control experiment is completed first to become familiar with the technology.
Steps in the sequencing workflow:
Prepare for your experiment
You will need to:
- Extract your DNA, and check its length, quantity and purity. The quality checks performed during the protocol are essential in ensuring experimental success.
- Ensure you have your sequencing kit, the correct equipment and third-party reagents
- Download the software for acquiring and analysing your data
- Check your flow cell to ensure it has enough pores for a good sequencing run
Library preparation
The table below is an overview of the steps required in the library preparation, including timings and optional stopping points.
Library preparation Process Time Stop option DNA repair and end-prep Repair the DNA and prepare the DNA ends for adapter attachment 35 minutes 4°C overnight Adapter ligation and clean-up Attach the sequencing adapters to the DNA ends 20 minutes 4°C short-term storage or for repeated use, such as re-loading your flow cell
-80°C for single-use, long-term storage.
We strongly recommend sequencing your library as soon as it is adapted.Priming and loading the flow cell Prime the flow cell and load the prepared library for sequencing 5 minutes 
Sequencing and analysis
You will need to:
- Start a sequencing run using the MinKNOW software which will collect raw data from the device and basecall reads.
- Start the EPI2ME software and select a bioinformatics workflow to analyse your data.

Equipment and consumables
- Materials
-
- 1 µg (or 100-200 fmol) high molecular weight genomic DNA
- OR 100+ ng high molecular weight genomic DNA if performing DNA fragmentation
- Ligation Sequencing Kit XL V14 (SQK-LSK114-XL)
- Consumables
-
- MinION and GridION Flow Cell
- NEBNext® Companion Module v2 for Oxford Nanopore Technologies® Ligation Sequencing (NEB, E7672S or E7672L)
- Agencourt AMPure XP beads (Beckman Coulter™ cat # A63881)
- 1.5 ml Eppendorf DNA LoBind tubes
- Eppendorf twin.tec® PCR plate 96 LoBind, semi-skirted (Eppendorf™, cat # 0030129504) with heat seals
- 0.2 ml thin-walled PCR tubes
- Nuclease-free water (e.g. ThermoFisher, AM9937)
- Freshly prepared 80% ethanol in nuclease-free water
- Qubit™ Assay Tubes (Invitrogen, Q32856)
- Qubit dsDNA HS Assay Kit (Invitrogen, Q32851)
- Bovine Serum Albumin (BSA) (50 mg/ml) (e.g Invitrogen™ UltraPure™ BSA 50 mg/ml, AM2616)
- Equipment
-
- MinION or GridION device
- MinION and GridION Flow Cell Light Shield
- Magnetic rack suitable for 96-well PCR plates, e.g. DynaMag™-96 Side Skirted Magnet (Thermo Fisher, cat # 12027)
- OR magnetic separator suitable for 0.2 ml PCR tube strips, e.g. DynaMag™-PCR Magnet (Thermo Fisher, #492025) or DynaMag™-96 Side Magnet (Thermo Fisher, #12331D)
- Hula mixer (gentle rotator mixer)
- Microfuge
- Microplate centrifuge, e.g. Fisherbrand™ Mini Plate Spinner Centrifuge (Fisher Scientific, 11766427)
- Vortex mixer
- Thermal cycler
- P1000 pipette and tips
- P200 pipette and tips
- P100 pipette and tips
- P20 pipette and tips
- P10 pipette and tips
- P2 pipette and tips
- Ice bucket with ice
- Timer
- Pipetting troughs
- Qubit fluorometer (or equivalent for QC check)
- Optional Equipment
-
- Agilent Bioanalyzer (or equivalent)
- Eppendorf 5424 centrifuge (or equivalent)
-
Adjust your sample input quantity depending on your initial DNA sample length:
Fragment library length Starting input Very short (<1 kb) 200 fmol Short (1-10 kb) 100–200 fmol Long (>10 kb) 1 µg For more information on sample input and flow cell loading amounts for our ligation sequencing protocols please visit our know-how document.
-
Input DNA
How to QC your input DNA
It is important that the input DNA meets the quantity and quality requirements. Using too little or too much DNA, or DNA of poor quality (e.g. highly fragmented or containing RNA or chemical contaminants) can affect your library preparation.
For instructions on how to perform quality control of your DNA sample, please read the Input DNA/RNA QC protocol.
Chemical contaminants
Depending on how the DNA is extracted from the raw sample, certain chemical contaminants may remain in the purified DNA, which can affect library preparation efficiency and sequencing quality. Read more about contaminants on the Contaminants page of the Community.
-
NEBNext® Companion Module v2 for Oxford Nanopore Technologies® Ligation Sequencing
We recommend buying the NEBNext® Companion Module v2 for Oxford Nanopore Technologies® Ligation Sequencing (catalogue number E7672S or E7672L), which contains all the NEB reagents needed for use with the Ligation Sequencing Kit.
The previous version, NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing (NEB, E7180S or E7180L) is compatible, but the recommended v2 module offers more efficient dA-tailing and ligation, a result of the FFPEv2 DNA Repair Buffer and Salt-T4 DNA Ligase, respectively. A marked cost saving per sample preparation is also realised when using the v2 module.
Note: for our amplicon protocols, NEBNext FFPE DNA Repair Mix is not required and purchasing the required reagents separately is more cost effective.
-
Multichannel pipettes
For scaling up library prep using the Ligation Sequencing Kit XL, customers will need multichannel pipettes and appropriate pipette tips. Although the choice of brand is left to the user's discretion, our R&D team can recommend Rainin Pipet-Lite LTS L200-XLS+ (20–200 μl) and Pipet-Lit LTS L20-XLS+ (2–20 μl) pipettes and Rainin LTS pipette tips.
-
Third-party reagents
We have validated and recommend the use of all the third-party reagents used in this protocol. Alternatives have not been tested by Oxford Nanopore Technologies.
For all third-party reagents, we recommend following the manufacturer's instructions to prepare the reagents for use.
-
Check your flow cell
We highly recommend that you check the number of pores in your flow cell prior to starting a sequencing experiment. This should be done within 12 weeks of purchasing for MinION/GridION/PromethION or within four weeks of purchasing Flongle Flow Cells. Oxford Nanopore Technologies will replace any flow cell with fewer than the number of pores in the table below, when the result is reported within two days of performing the flow cell check, and when the storage recommendations have been followed. To do the flow cell check, please follow the instructions in the Flow Cell Check document.
Flow cell Minimum number of active pores covered by warranty Flongle Flow Cell 50 MinION/GridION Flow Cell 800 PromethION Flow Cell 5000 -
Ligation Sequencing Kit XL V14 (SQK-LSK114-XL) contents

Name Acronym Vial colour Number of vials Fill volume per vial (µl) DNA Control Strand DCS Yellow 1 100 Ligation Adapter LA Green 1 320 Ligation Buffer LNB White 1 1,500 Elution Buffer EB White cap, black strip label 1 10,000 Long Fragment Buffer LFB White cap, orange strip label 2 20,000 Short Fragment Buffer SFB White cap, blue strip label 2 20,000 Library Beads LIB Pink 2 1,800 Library Solution LIS White cap, pink label 2 1,800 Sequencing Buffer SB Red 3 1,700 Flow Cell Flush FCF Clear 4 15,500 Flow Cell Tether FCT Purple 1 1,600 Note: The DNA Control Sample (DCS) is a 3.6 kb standard amplicon mapping the 3' end of the Lambda genome.

Library preparation
DNA repair and end-prep
DNA repair and end-prep
- Materials
-
- 1 µg (or 100-200 fmol) high molecular weight genomic DNA
- DNA Control Sample (DCS)
- Consumables
-
- NEBNext® FFPE DNA Repair Mix (M6630) from the NEBNext® Companion Module v2 (NEB, E7672S or E7672L)
- NEBNext® Ultra II End Prep Enzyme Mix (E7646) from the NEBNext® Companion Module v2 (NEB, E7672S or E7672L)
- NEBNext® FFPE DNA Repair Buffer v2 (E7363) from the NEBNext® Companion Module v2 (NEB, E7672S or E7672L)
- Eppendorf twin.tec® PCR plate 96 LoBind, semi-skirted (Eppendorf™, cat # 0030129504) with heat seals
- OR 0.2 ml thin-walled PCR tubes
- Agencourt AMPure XP Beads (Beckman Coulter™, A63881)
- Nuclease-free water (e.g. ThermoFisher, AM9937)
- Freshly prepared 80% ethanol in nuclease-free water
- Qubit™ Assay Tubes (Invitrogen, Q32856)
- Qubit dsDNA HS Assay Kit (Invitrogen, Q32851)
- Equipment
-
- Magnetic rack suitable for 96-well PCR plates, e.g. DynaMag™-96 Side Skirted Magnet (Thermo Fisher, cat # 12027)
- OR magnetic separator suitable for 0.2 ml PCR tube strips, e.g. DynaMag™-PCR Magnet (Thermo Fisher, #492025) or DynaMag™-96 Side Magnet (Thermo Fisher, #12331D)
- P1000 pipette and tips
- P100 pipette and tips
- P10 pipette and tips
- Thermal cycler
- Microfuge
- Microplate centrifuge, e.g. Fisherbrand™ Mini Plate Spinner Centrifuge (Fisher Scientific, 11766427)
- Vortex mixer
- Ice bucket with ice
- Pipetting troughs
- Qubit fluorometer (or equivalent for QC check)
-
Check your flow cell.
We recommend performing a flow cell check before starting your library prep to ensure you have a flow cell with enough pores for a good sequencing run.
See the flow cell check instructions in the MinKNOW protocol for more information.
-
Thaw DNA Control Sample (DCS) at room temperature, spin down, mix by pipetting, and place on ice.
-
Prepare the NEB reagents in accordance with manufacturer’s instructions, and place on ice.
For optimal performance, NEB recommend the following:
Thaw all reagents on ice.
Flick and/or invert the reagent tubes to ensure they are well mixed.
Note: Do not vortex the FFPE DNA Repair Mix or Ultra II End Prep Enzyme Mix.Always spin down tubes before opening for the first time each day.
Vortex the FFPE DNA Repair Buffer v2, or the NEBNext FFPE DNA Repair Buffer and Ultra II End Prep Reaction Buffer to ensure they are well mixed.
Note: These buffers may contain a white precipitate. If this occurs, allow the mixture(s) to come to room temperature and pipette the buffer several times to break up the precipitate, followed by a quick vortex to mix.The FFPE DNA Repair Buffer may have a yellow tinge and is fine to use if yellow.
-
Prepare the DNA in nuclease-free water:
- Transfer 1 μg (or 100-200 fmol) input DNA into a separate well of a 96-well plate or a 0.2 ml PCR tube strip
- Adjust the volume to 47 μl with nuclease-free water
- Mix thoroughly by pipetting up and down, or by flicking the tube
- If necessary, seal and spin down briefly in an appropriate microfuge
-
To each sample, add the following:
Between each addition, pipette mix 10-20 times.
Reagent Volume DNA from the previous step 47 µl DNA CS (optional) 1 µl NEBNext FFPE DNA Repair Buffer v2 7 µl NEBNext FFPE DNA Repair Mix 2 µl Ultra II End-prep Enzyme Mix 3 µl Total 60 µl If using the previous version of the NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing (NEB, E7180S or E7180L):
Between each addition, pipette mix 10-20 times.
Reagent Volume DNA from the previous step 47 µl DNA CS (optional) 1 µl NEBNext FFPE DNA Repair Buffer 3.5 µl NEBNext FFPE DNA Repair Mix 2 µl Ultra II End-prep Reaction Buffer 3.5 µl Ultra II End-prep Enzyme Mix 3 µl Total 60 µl -
Mix well by gently pipetting the entire volume within each well/tube up and down 10 times, or by flicking the tubes, and spin down.
-
Seal the plate, or close the tube lids.
-
Using a thermal cycler, incubate at 20°C for 5 minutes and 65°C for 5 minutes.
-
Resuspend the AMPure XP beads by vortexing and transfer to a pipetting trough. Ensure that the volume transferred is enough for 60 µl to be added to each DNA sample, with an excess to allow for dead volume within the pipetting trough.
-
Keep the DNA samples in their original wells/PCR tubes. Add 60 µl of resuspended AMPure XP beads to each sample and mix by pipetting at least 100 µl up and down ten times. Retain any unused beads.
-
Incubate for 5 minutes at room temperature.
-
Prepare fresh 80% ethanol in nuclease-free water and pour into a pipetting trough. Allow enough for 500 µl per sample, with an excess to allow for dead volume within the pipetting trough. After the bead washing steps, discard any unused ethanol.
-
Pellet the beads on a magnet for at least 2 minutes, or until the supernatant is clear. Keep the plate/tube strip on the magnet and pipette off the supernatant.
-
Keeping the plate/tube strip on the magnet, wash each pellet of beads with 200 µl of the freshly-prepared 80% ethanol without disturbing the pellets. Remove the 80% ethanol using a pipette and discard.
-
Repeat the previous step.
-
Seal the plate, or close the tube lids. Spin down and place the plate/tube strip back on the magnet. Pipette off any residual ethanol.
-
Pour nuclease-free water into a pipetting trough. Allow enough for 61 µl per sample, with an excess to allow for dead volume within the pipetting trough.
-
Remove the plate/tube strip from the magnetic rack and resuspend each pellet in 61 µl nuclease-free water from the pipetting trough. Pipette the entire volume up and down ten times).
-
Seal the plate or close the tube lids, and incubate for 2 minutes at room temperature.
-
Pellet the beads on a magnet until the eluate is clear and colourless, for at least 1 minute.
-
Remove and retain 61 µl of each eluate in a separate, clean well/tube within a 96-well PCR plate or PCR tube strip. Dispose of the pelleted beads.
-
Quantify 1 µl of eluted sample using a Qubit fluorometer.
Adapter ligation and clean-up
Adapter ligation and clean-up
- Materials
-
- Ligation Adapter (LA)
- Ligation Buffer (LNB)
- Long Fragment Buffer (LFB)
- Short Fragment Buffer (SFB)
- Elution Buffer (EB)
- Consumables
-
- Salt-T4® DNA Ligase (NEB, M0467)
- Agencourt AMPure XP Beads (Beckman Coulter™, A63881)
- 1.5 ml Eppendorf DNA LoBind tubes
- Qubit™ Assay Tubes (Invitrogen, Q32856)
- Qubit dsDNA HS Assay Kit (Invitrogen, Q32851)
- Equipment
-
- Magnetic rack suitable for 96-well PCR plates, e.g. DynaMag™-96 Side Skirted Magnet (Thermo Fisher, cat # 12027)
- OR magnetic separator suitable for 0.2 ml PCR tube strips, e.g. DynaMag™-PCR Magnet (Thermo Fisher, #492025) or DynaMag™-96 Side Magnet (Thermo Fisher, #12331D)
- Microfuge
- Microplate centrifuge, e.g. Fisherbrand™ Mini Plate Spinner Centrifuge (Fisher Scientific, 11766427)
- Vortex mixer
- Multichannel pipettes suitable for dispensing 2–20 μl and 20–200 μl, and tips
- P1000 pipette and tips
- P100 pipette and tips
- P20 pipette and tips
- P10 pipette and tips
- Pipetting troughs
- Qubit fluorometer (or equivalent for QC check)
-
Spin down the Ligation Adapter (LA) and Salt-T4® DNA Ligase, and place on ice.
-
Thaw Ligation Buffer (LNB) at room temperature, spin down and mix by pipetting. Due to viscosity, vortexing this buffer is ineffective. Place on ice immediately after thawing and mixing.
-
Thaw the Elution Buffer (EB) at room temperature and mix by vortexing. Then spin down and place on ice.
-
Thaw either Long Fragment Buffer (LFB) or Short Fragment Buffer (SFB) at room temperature and mix by vortexing. Then spin down and keep at room temperature.
-
For each sample, combine the following reagents:
Between each addition, pipette mix 10-20 times.
Reagent Volume DNA sample from the previous step 60 µl Ligation Adapter (LA) 5 µl Ligation Buffer (LNB) 25 µl Salt-T4® DNA Ligase 10 µl Total 100 µl -
Mix well by gently pipetting the entire volume within each well/tube up and down 10 times.
-
Incubate the reaction for 10 minutes at room temperature.
-
Resuspend the AMPure XP beads by vortexing and transfer to a pipetting trough. Ensure that the volume transferred is enough for 60 µl to be added to each DNA sample, with an excess to allow for dead volume within the pipetting trough.
-
Add 40 µl of resuspended AMPure XP beads to each sample and mix by pipetting the entire combined volume up and down 10 times.
-
Incubate for 5 minutes at room temperature.
-
Add sufficient Long Fragment Buffer (LFB) or Short Fragment Buffer (SFB) to a pipetting trough. Allow enough for 400 µl per sample, with an excess to allow for dead volume within the pipetting trough. Retain any unused reagent after the wash steps.
-
Pellet the beads on a magnet for at least 2 minutes, or until the supernatant is clear. Keep the plate/tube strip on the magnet and pipette off the supernatant.
-
Remove the plate/tube strip from the magnetic rack and wash each pellet of beads by adding either 200 μl Long Fragment Buffer (LFB) or Short Fragment Buffer (SFB). Resuspend each pellet thoroughly by pipetting the entire volume of buffer up and down ten times. Fully resuspending the beads at this step ensures optimal kit performance. Return the plate/tube strip to the magnetic rack and allow the beads to pellet until the supernatant is clear. Remove the supernatant using a pipette and discard.
-
Repeat the previous step.
-
Seal the plate, or close the tube lids. Spin down and place the plate/tube strip back on the magnet. Pipette off any residual supernatant.
-
Add sufficient Elution Buffer (EB) to a pipetting trough. Allow enough for 15 µl per sample, with an excess to allow for dead volume within the pipetting trough. Retain any unused Elution Buffer (EB) after the elution step.
-
Remove the plate/tube strip from the magnetic rack and resuspend each pellet in 15 µl Elution Buffer (EB) from the pipetting trough, pipetting the entire volume up and down 10 times.
-
Seal the plate (or close the tube lids), and incubate for 10 minutes at 37°C in a thermal cycler. Any heated lid used should be limited to 50°C.
-
Pellet the beads on a magnet until the eluate is clear and colourless, for at least 1 minute.
-
Remove and retain 15 µl of each eluate in a separate, clean well/tube within a 96-well PCR plate or PCR tube strip. Dispose of the pelleted beads.
-
Quantify 1 µl of eluted sample using a Qubit fluorometer.
-
Depending on your DNA library fragment size, prepare your final library in 12 µl of Elution Buffer (EB).
Fragment library length Flow cell loading amount Very short (<1 kb) 100 fmol Short (1-10 kb) 35–50 fmol Long (>10 kb) 300 ng Note: If the library yields are below the input recommendations, load the entire library.
If required, we recommend using a mass to mol calculator such as the NEB calculator.
Priming and loading the MinION and GridION Flow Cell
Priming and loading the MinION and GridION Flow Cell
- Materials
-
- Flow Cell Flush (FCF)
- Flow Cell Tether (FCT)
- Library Solution (LIS)
- Library Beads (LIB)
- Sequencing Buffer (SB)
- Consumables
-
- MinION and GridION Flow Cell
- 1.5 ml Eppendorf DNA LoBind tubes
- Nuclease-free water (e.g. ThermoFisher, AM9937)
- Bovine Serum Albumin (BSA) (50 mg/ml) (e.g Invitrogen™ UltraPure™ BSA 50 mg/ml, AM2616)
- Equipment
-
- MinION or GridION device
- MinION and GridION Flow Cell Light Shield
- P1000 pipette and tips
- P100 pipette and tips
- P20 pipette and tips
- P10 pipette and tips
-
Thaw the Sequencing Buffer (SB), Library Beads (LIB) or Library Solution (LIS, if using), Flow Cell Tether (FCT) and Flow Cell Flush (FCF) at room temperature before mixing by vortexing. Then spin down and store on ice.
-
To prepare the flow cell priming mix with BSA, combine Flow Cell Flush (FCF) and Flow Cell Tether (FCT), as directed below. Mix by pipetting at room temperature.
Note: We are in the process of reformatting our kits with single-use tubes into a bottle format. Please follow the instructions for your kit format.
Single-use tubes format:
Add 5 µl Bovine Serum Albumin (BSA) at 50 mg/ml and 30 µl Flow Cell Tether (FCT) directly to a tube of Flow Cell Flush (FCF).Bottle format:
In a suitable tube for the number of flow cells, combine the following reagents:Reagent Volume per flow cell Flow Cell Flush (FCF) 1,170 µl Bovine Serum Albumin (BSA) at 50 mg/ml 5 µl Flow Cell Tether (FCT) 30 µl Total volume 1,205 µl -
Open the MinION or GridION device lid and slide the flow cell under the clip. Press down firmly on the priming port cover to ensure correct thermal and electrical contact.


-
Optional ActionComplete a flow cell check to assess the number of pores available before loading the library.
This step can be omitted if the flow cell has been checked previously.
See the flow cell check instructions in the MinKNOW protocol for more information.
-
Slide the flow cell priming port cover clockwise to open the priming port.

-
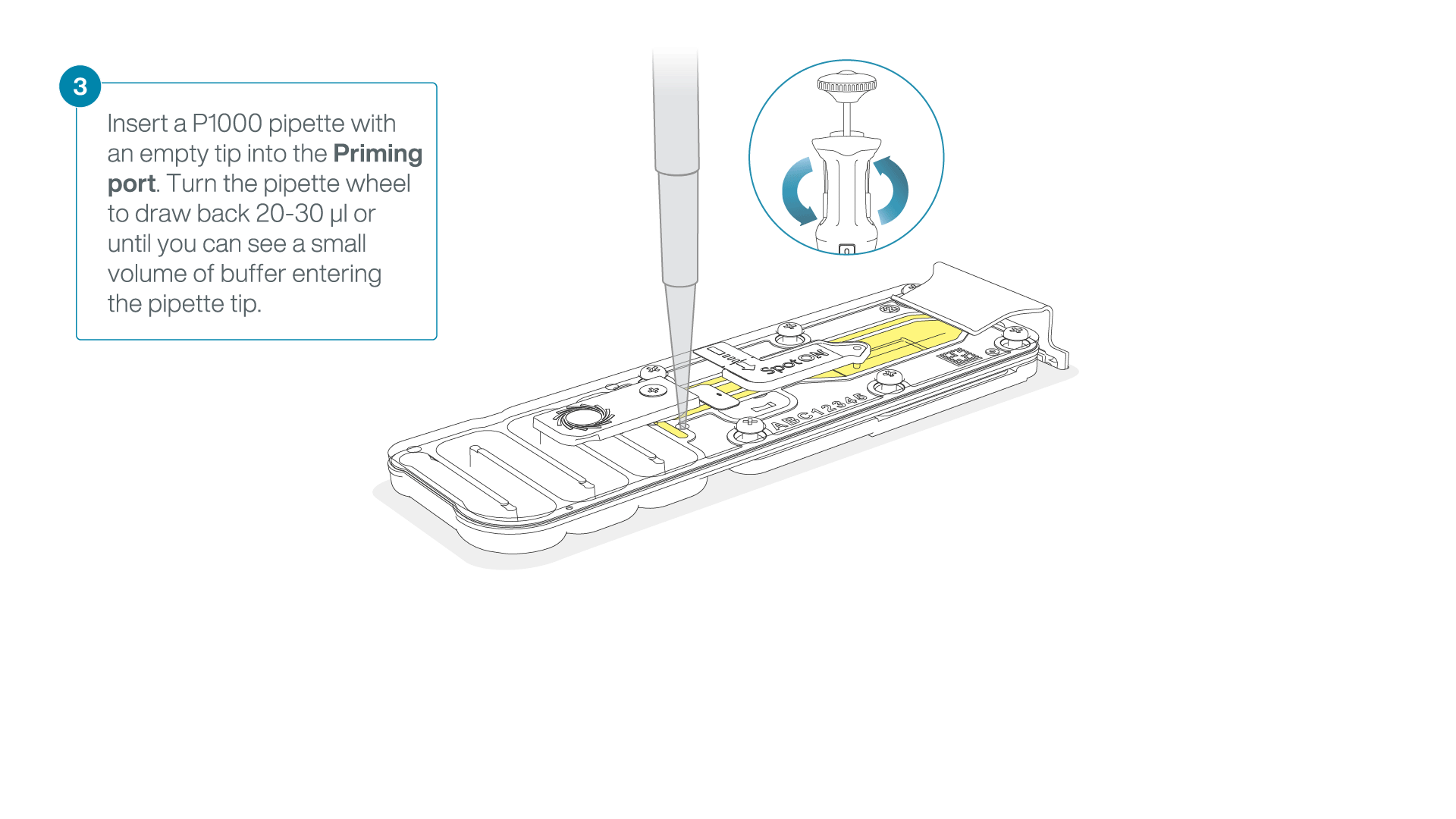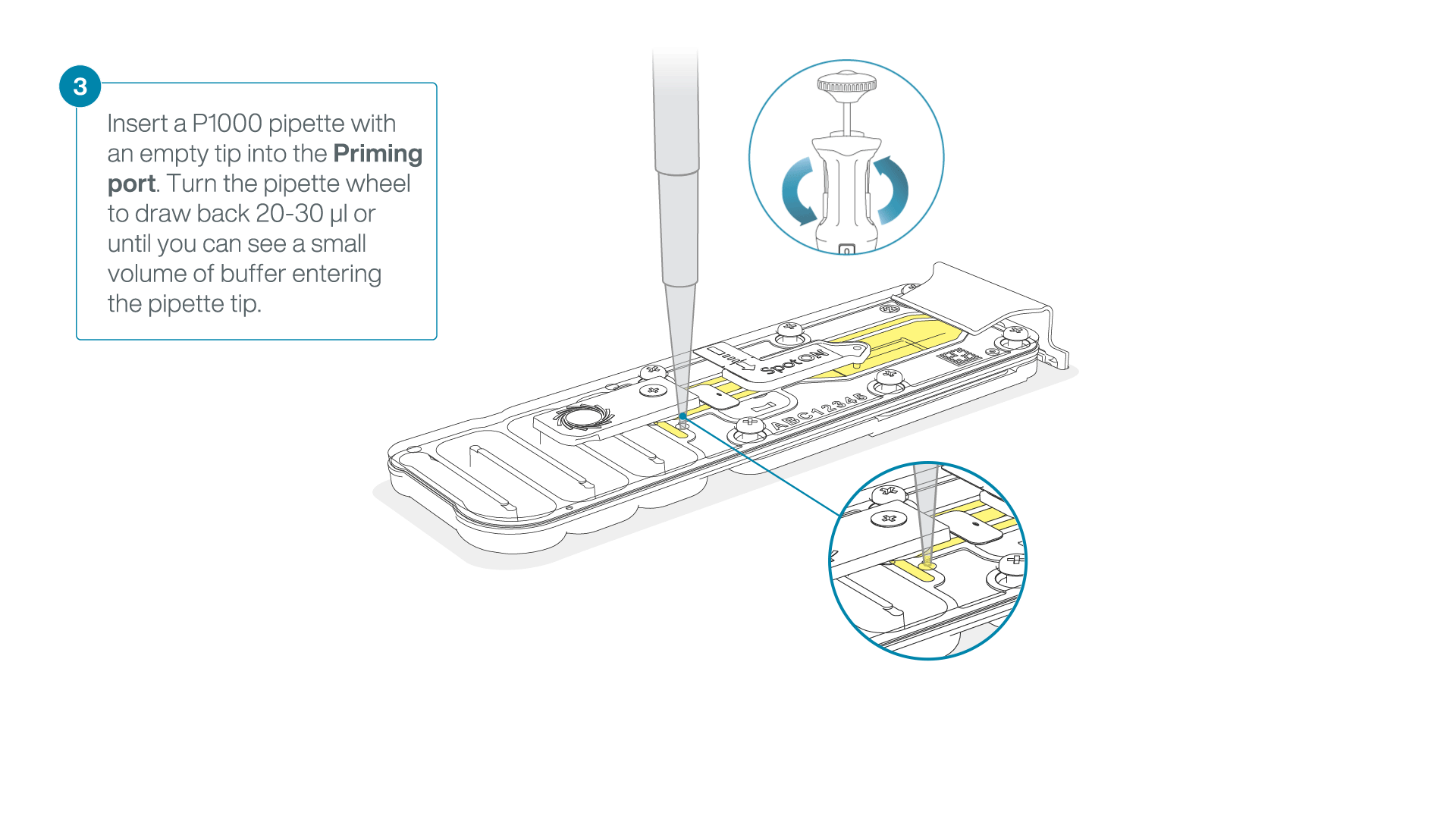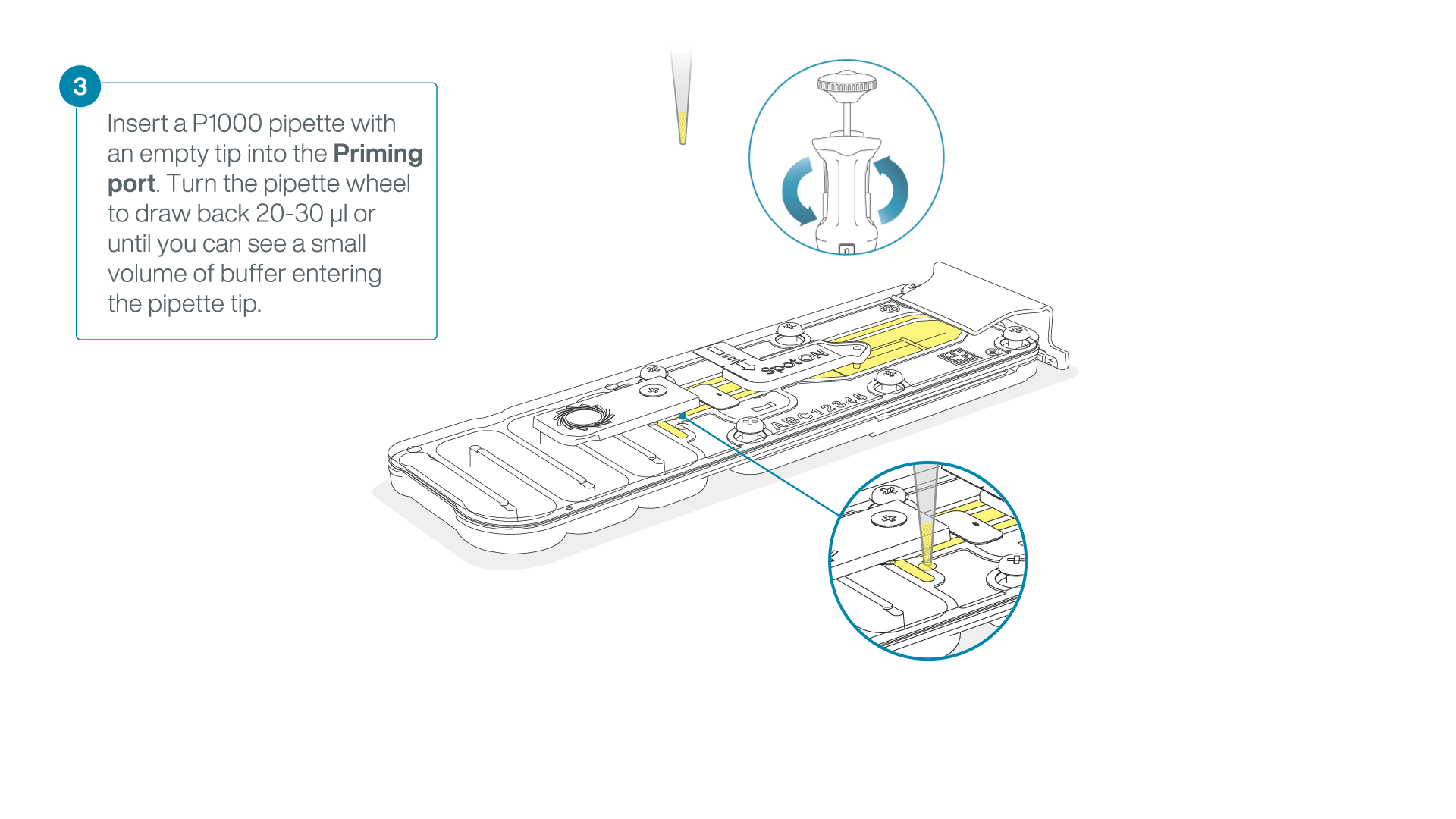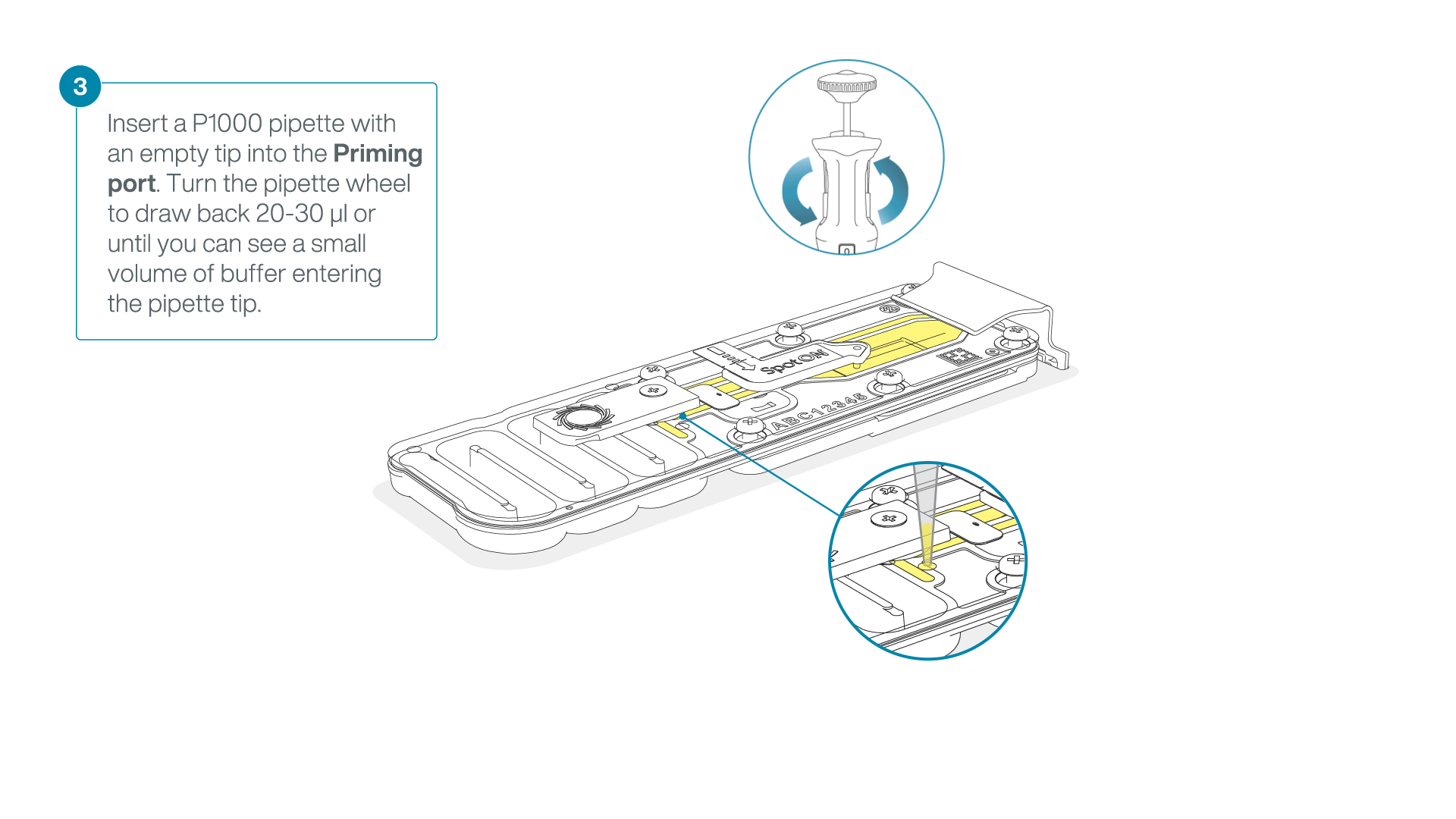
After opening the priming port, check for a small air bubble under the cover. Draw back a small volume to remove any bubbles:
- Set a P1000 pipette to 200 µl
- Insert the tip into the priming port
- Turn the wheel until the dial shows 220-230 µl, to draw back 20-30 µl, or until you can see a small volume of buffer entering the pipette tip
Note: Visually check that there is continuous buffer from the priming port across the sensor array.
-
Load 800 µl of the priming mix into the flow cell via the priming port, avoiding the introduction of air bubbles. Wait for five minutes. During this time, prepare the library for loading by following the steps below.
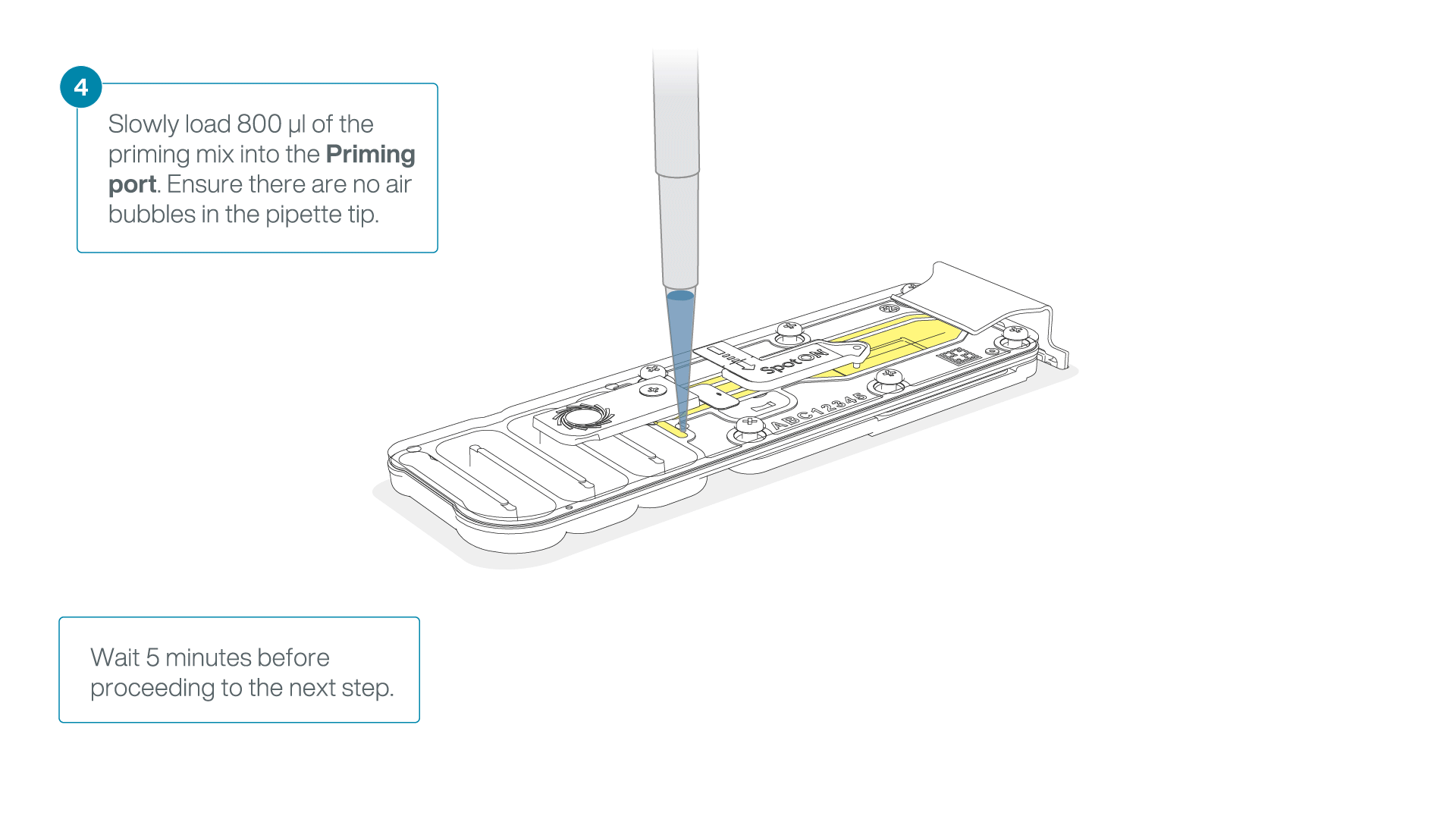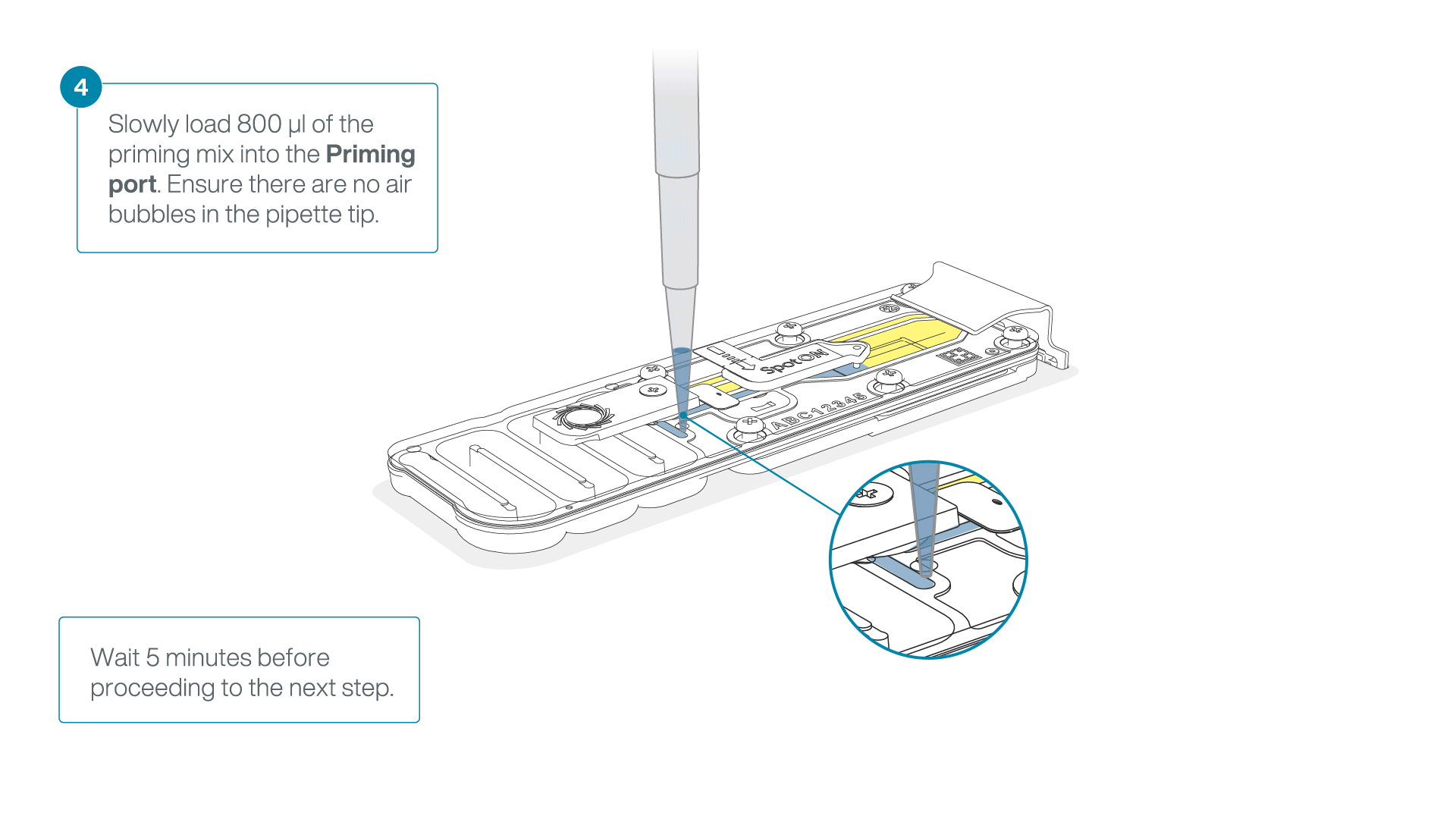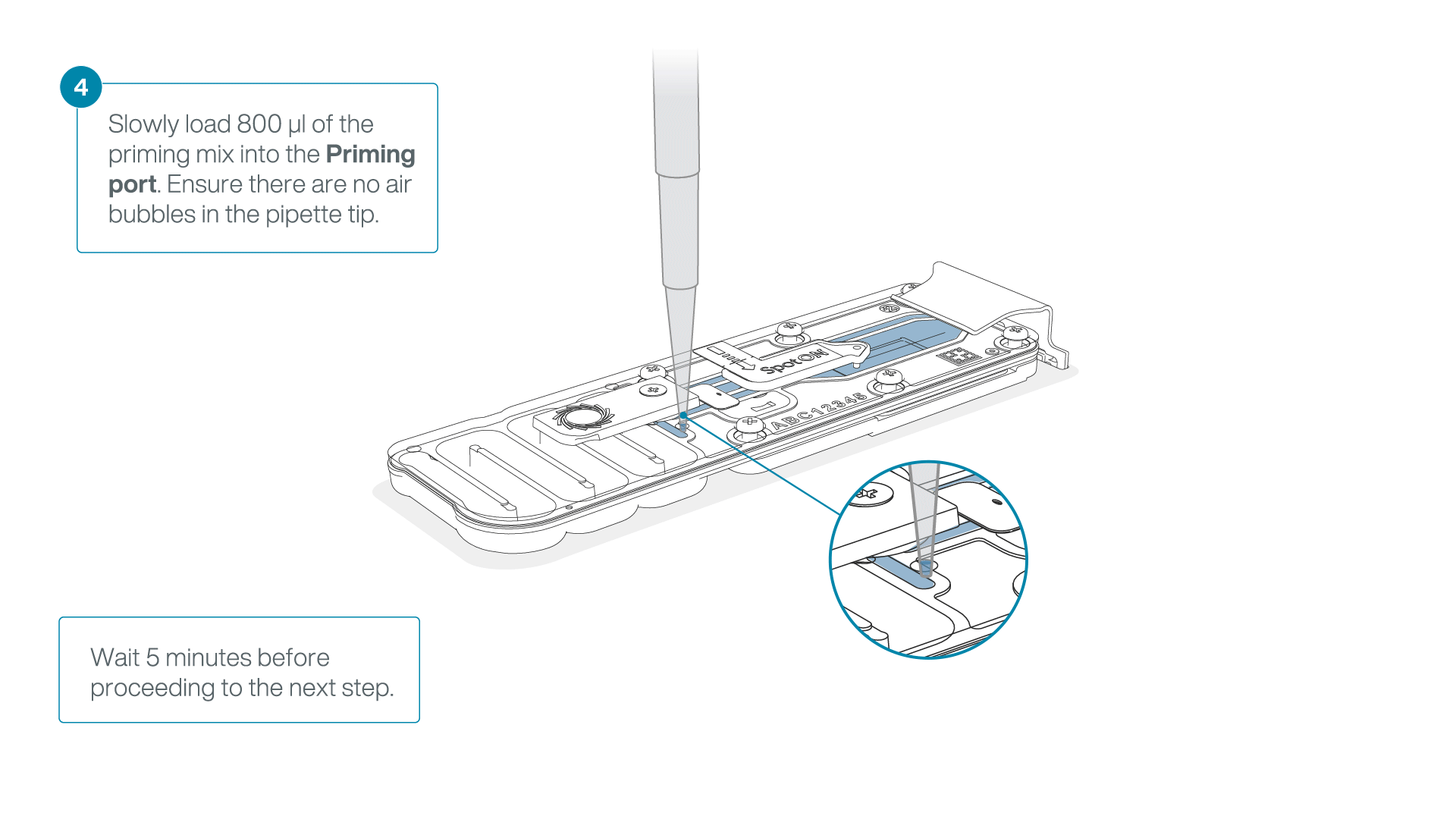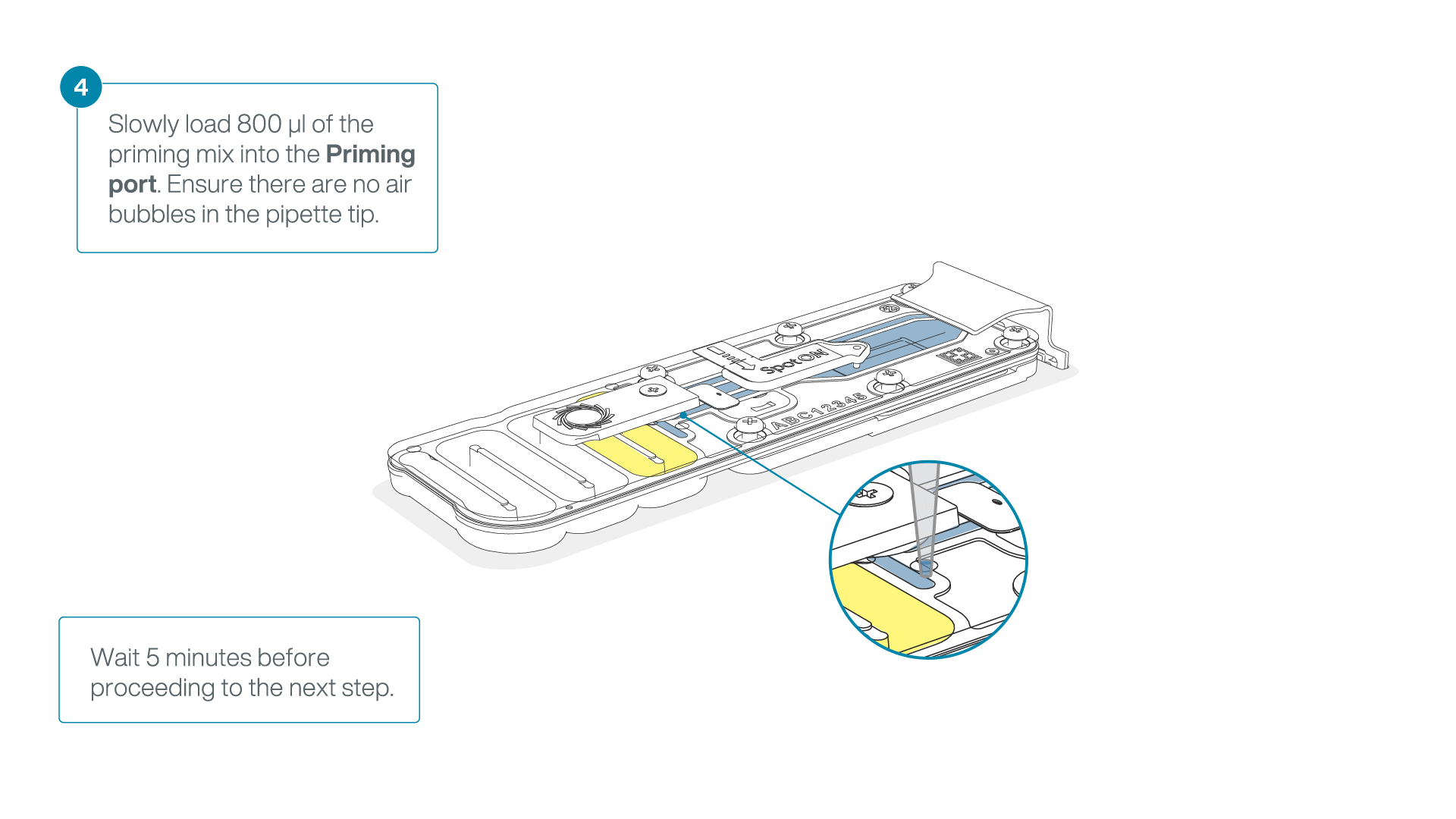
-
Thoroughly mix the contents of the Library Beads (LIB) by pipetting.
-
In a new 1.5 ml Eppendorf DNA LoBind tube, prepare the library for loading as follows:
Reagent Volume per flow cell Sequencing Buffer (SB) 37.5 µl Library Beads (LIB) mixed immediately before use, or Library Solution (LIS), if using 25.5 µl DNA library 12 µl Total 75 µl -
Complete the flow cell priming:
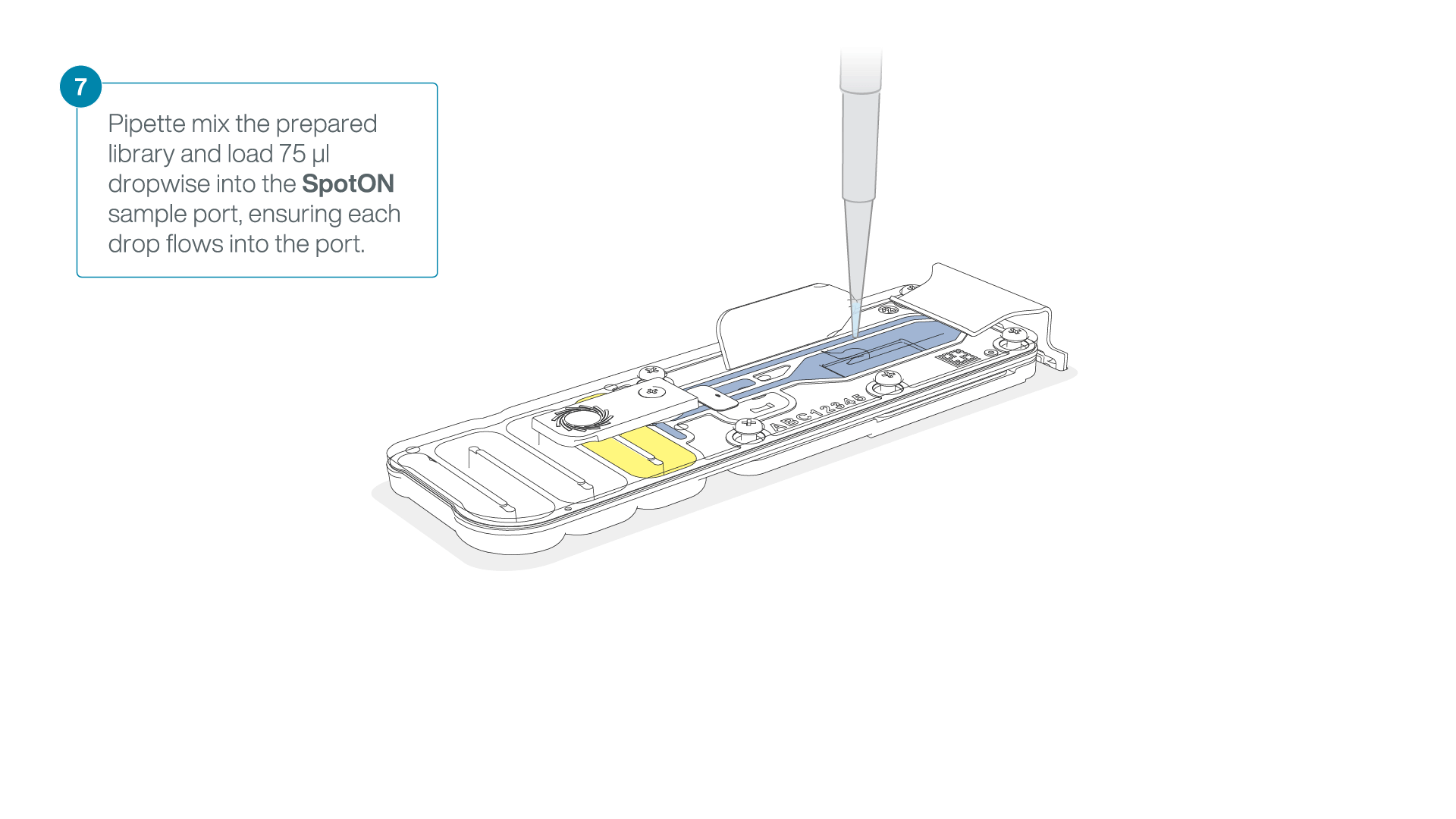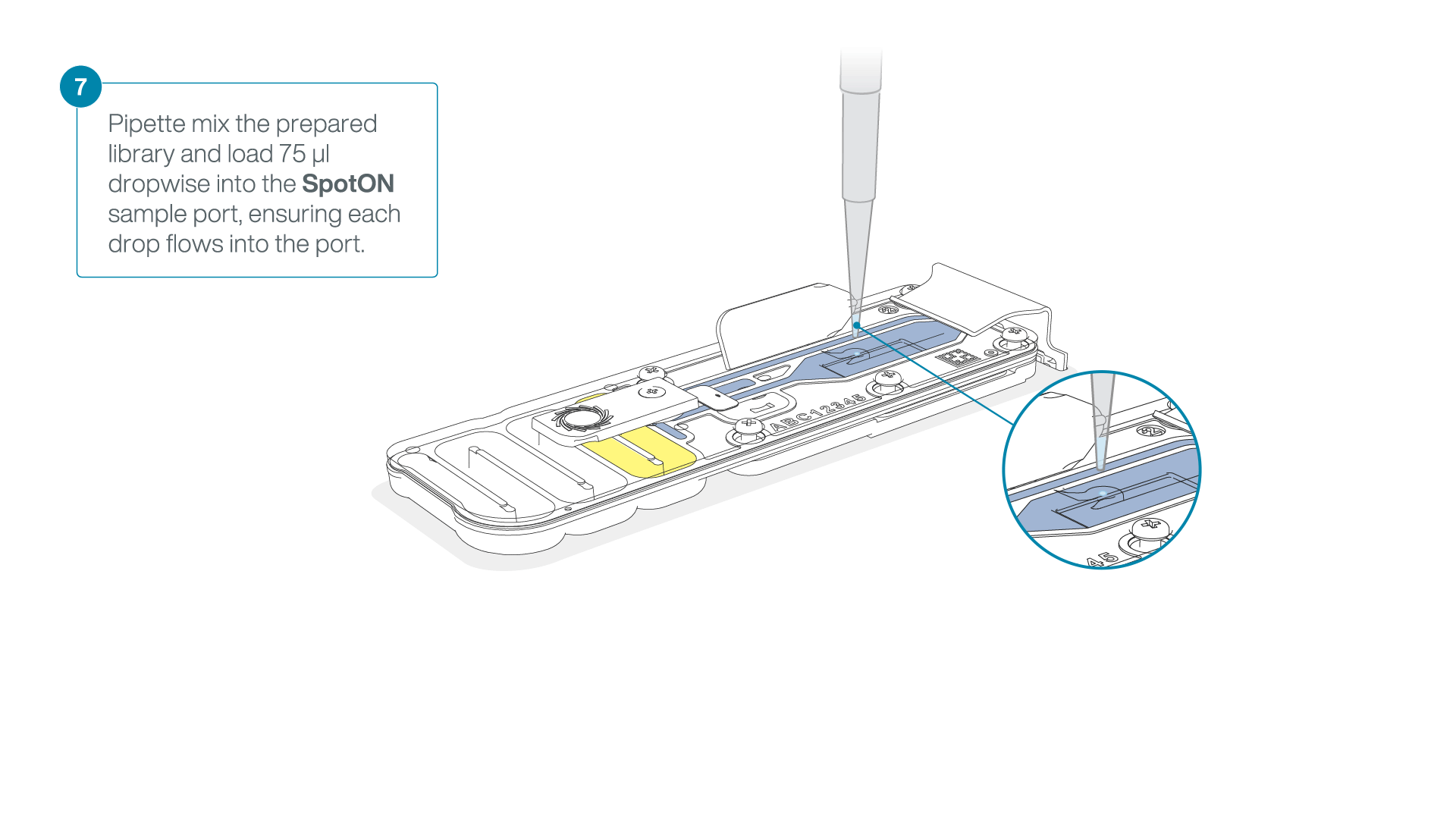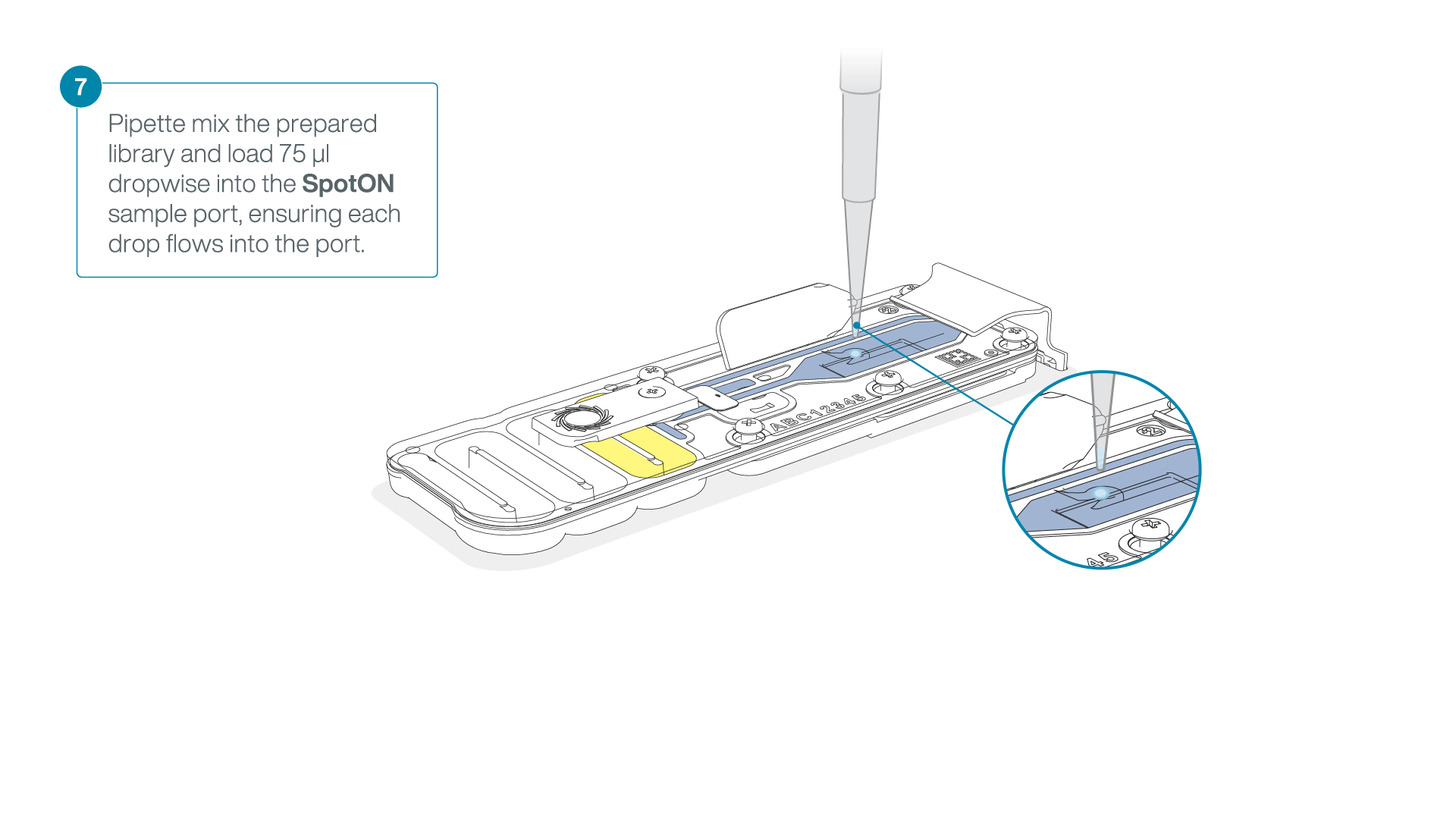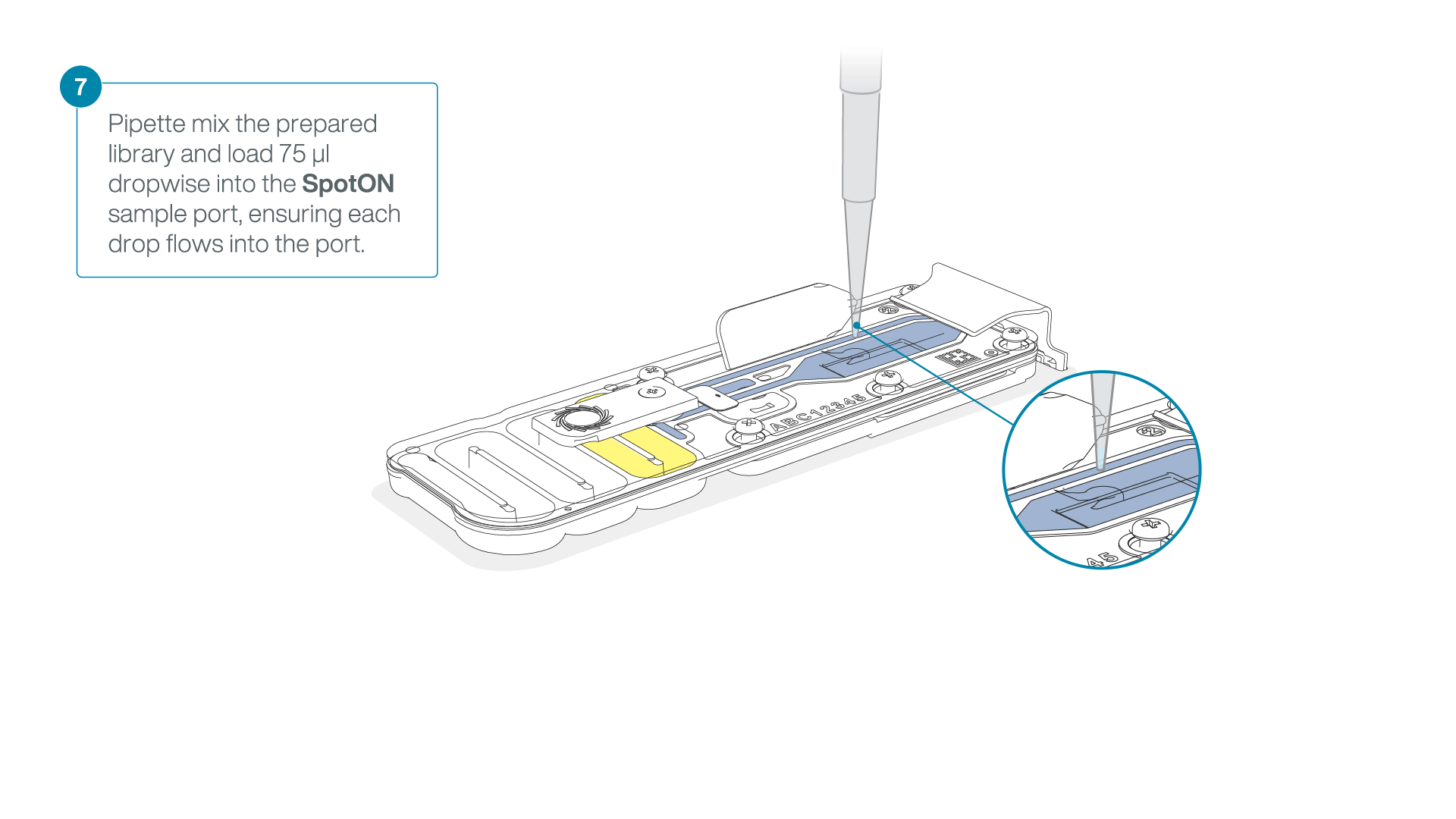
- Gently lift the SpotON sample port cover to make the SpotON sample port accessible.
- Load 200 µl of the priming mix into the flow cell priming port (not the SpotON sample port), avoiding the introduction of air bubbles.

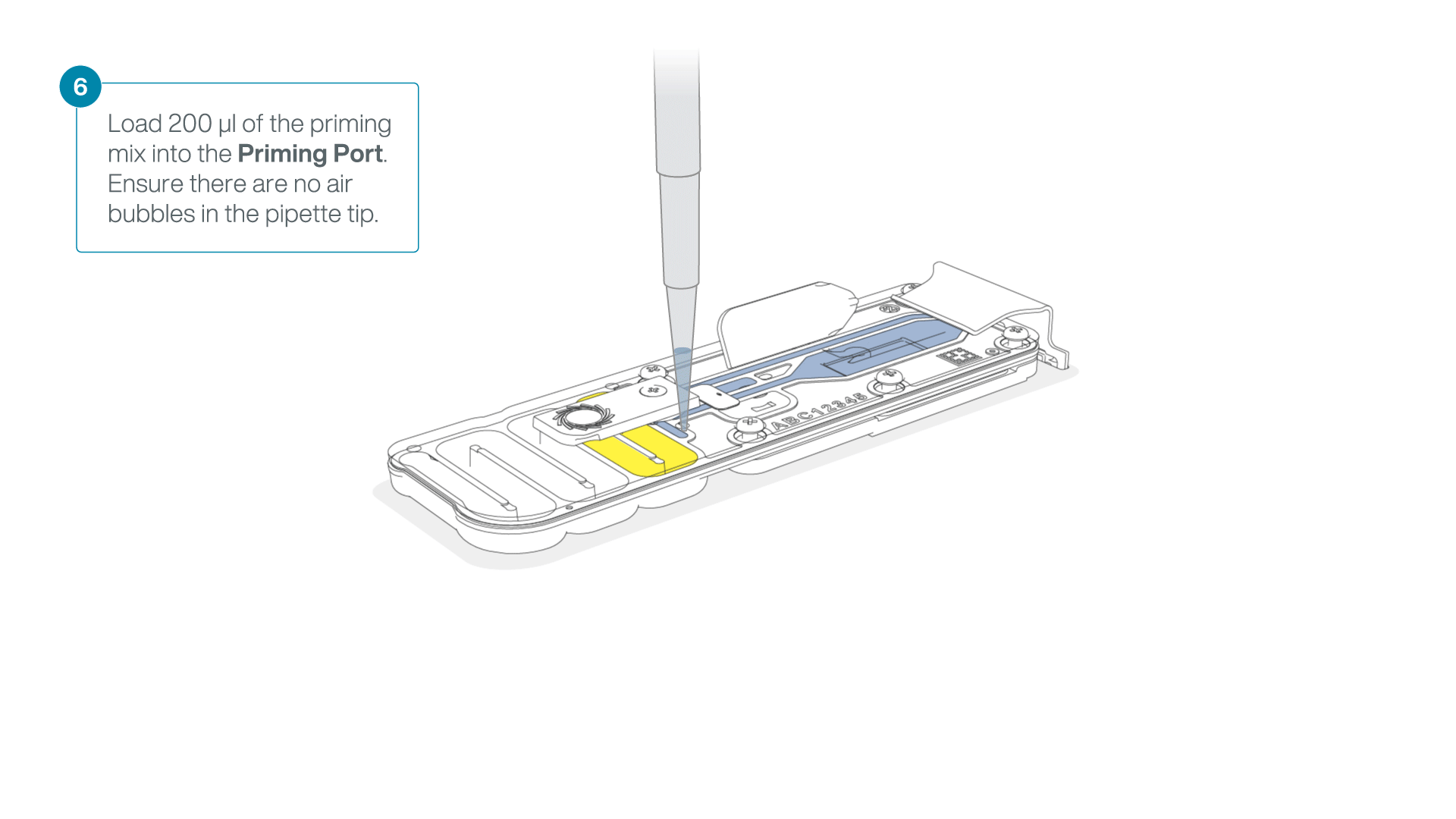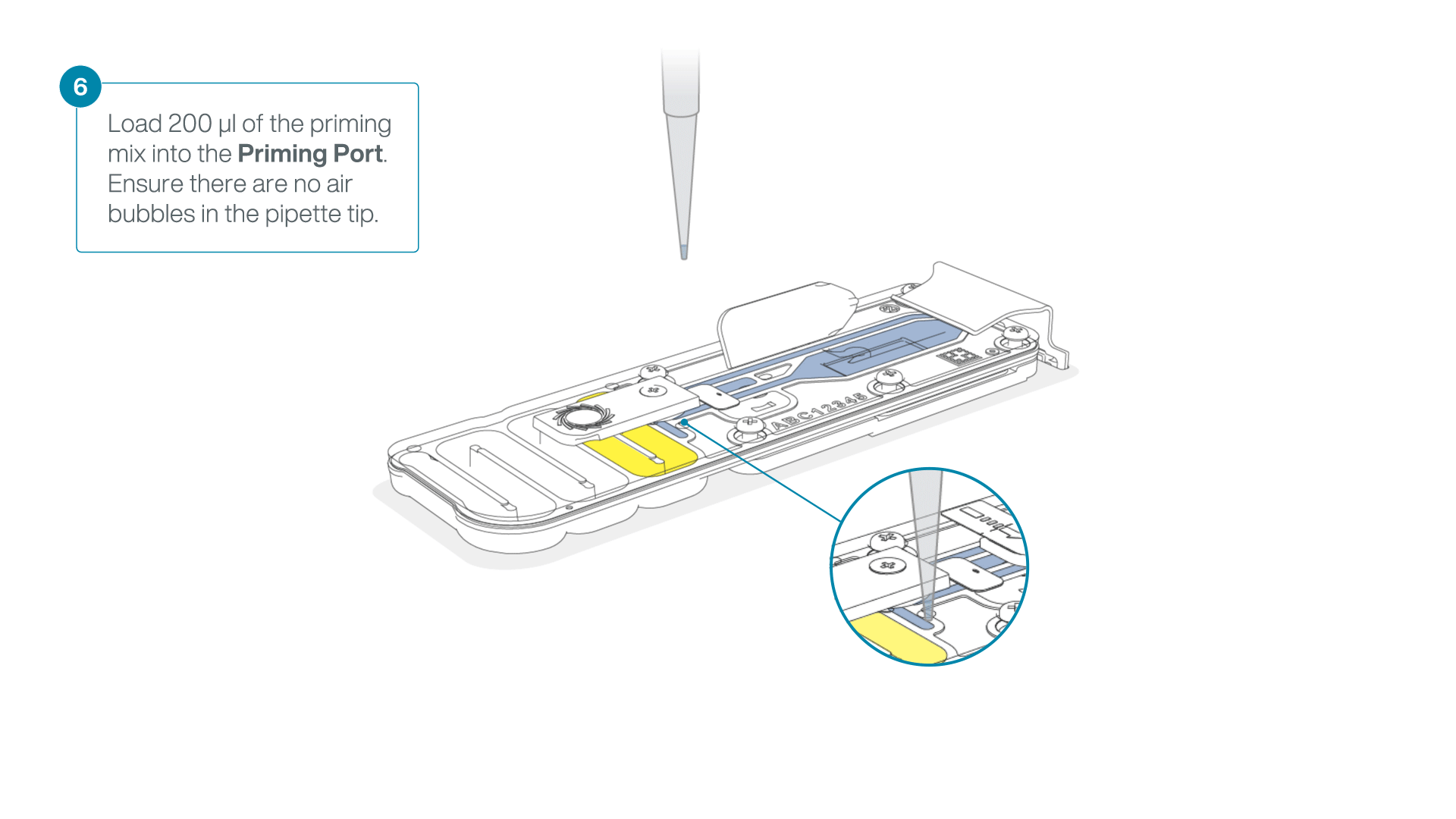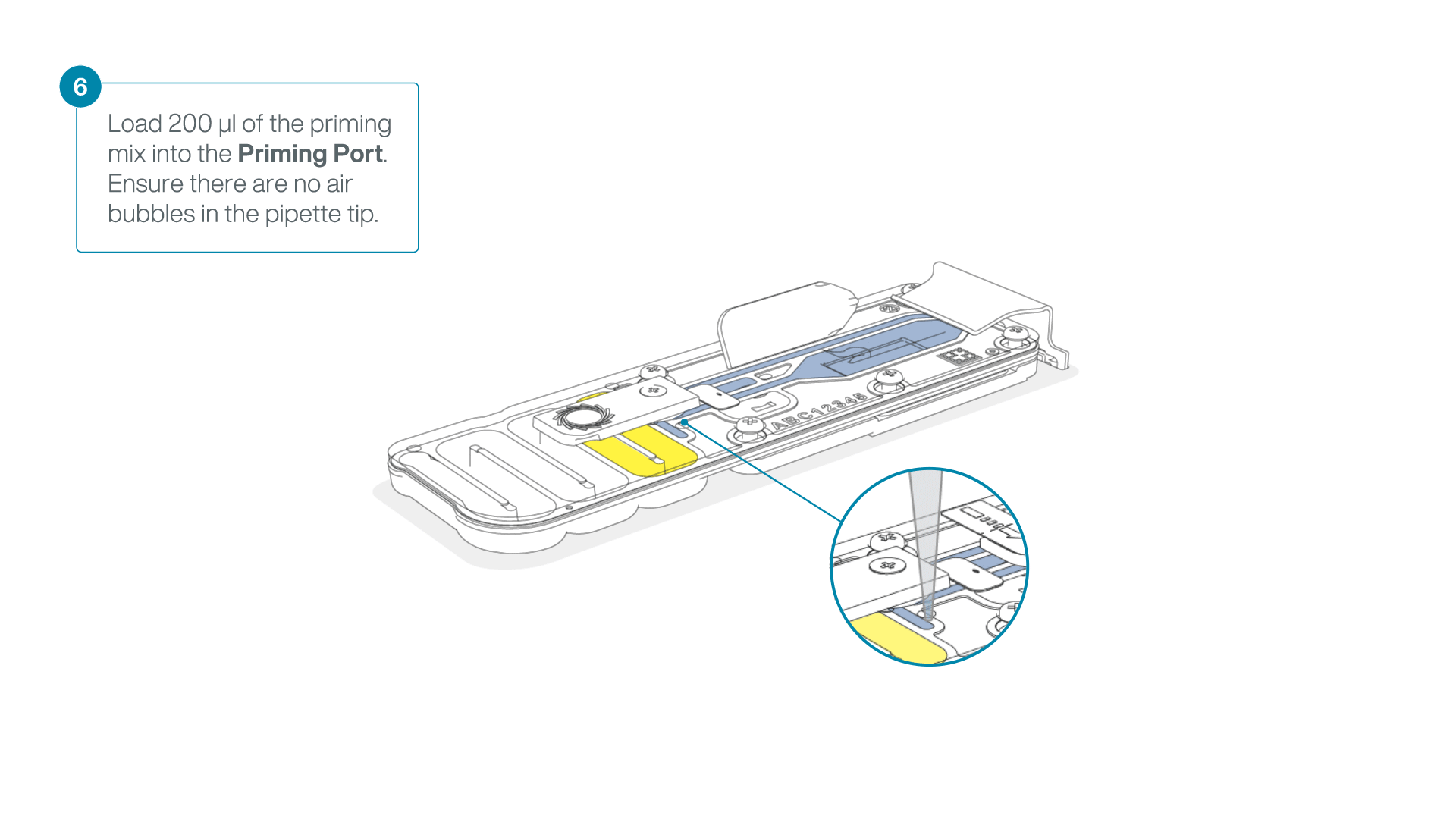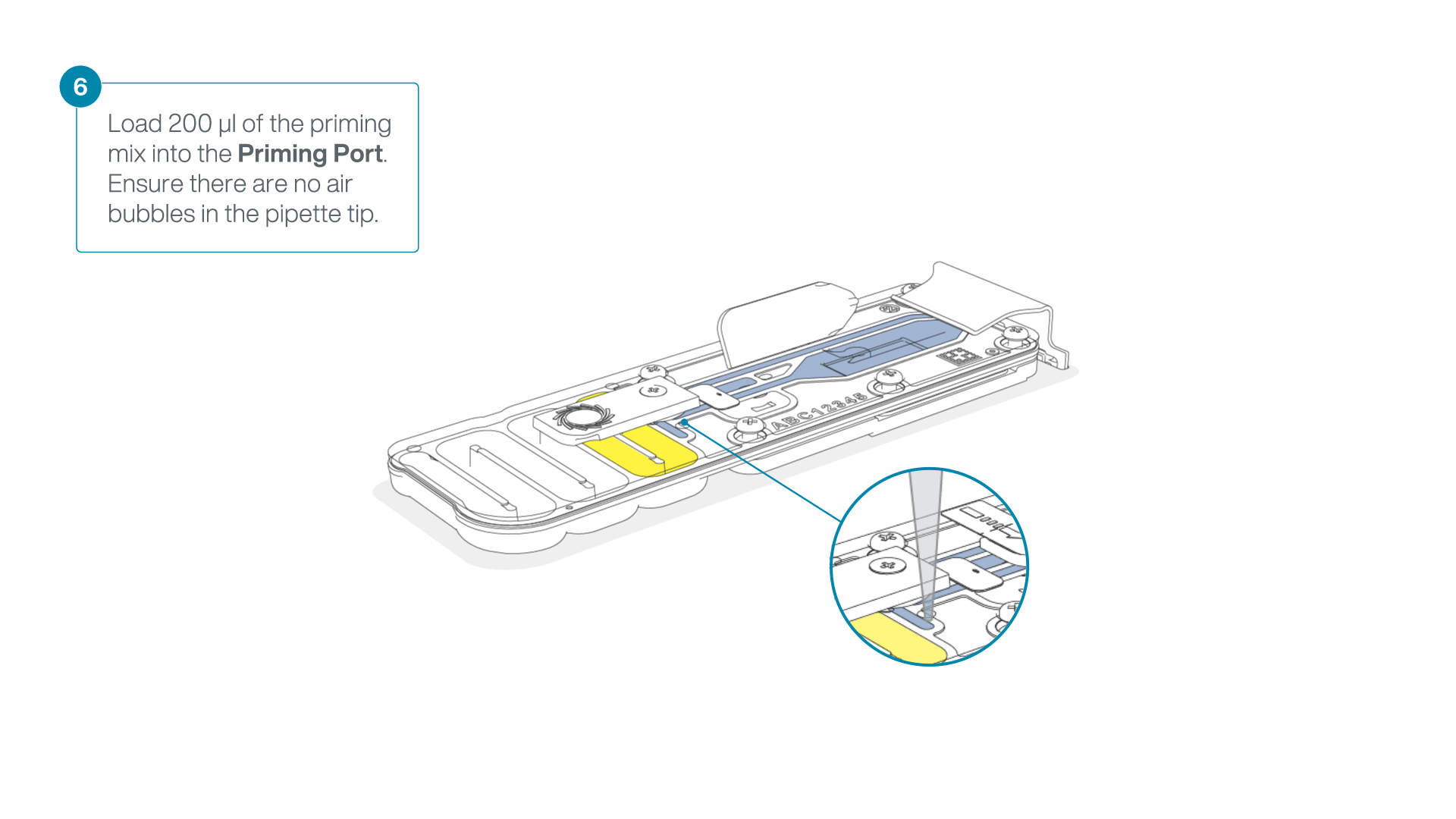
-
Mix the prepared library gently by pipetting up and down just prior to loading.
-
Add 75 μl of the prepared library to the flow cell via the SpotON sample port in a dropwise fashion. Ensure each drop flows into the port before adding the next.
-
Gently replace the SpotON sample port cover, making sure the bung enters the SpotON port and close the priming port.


-
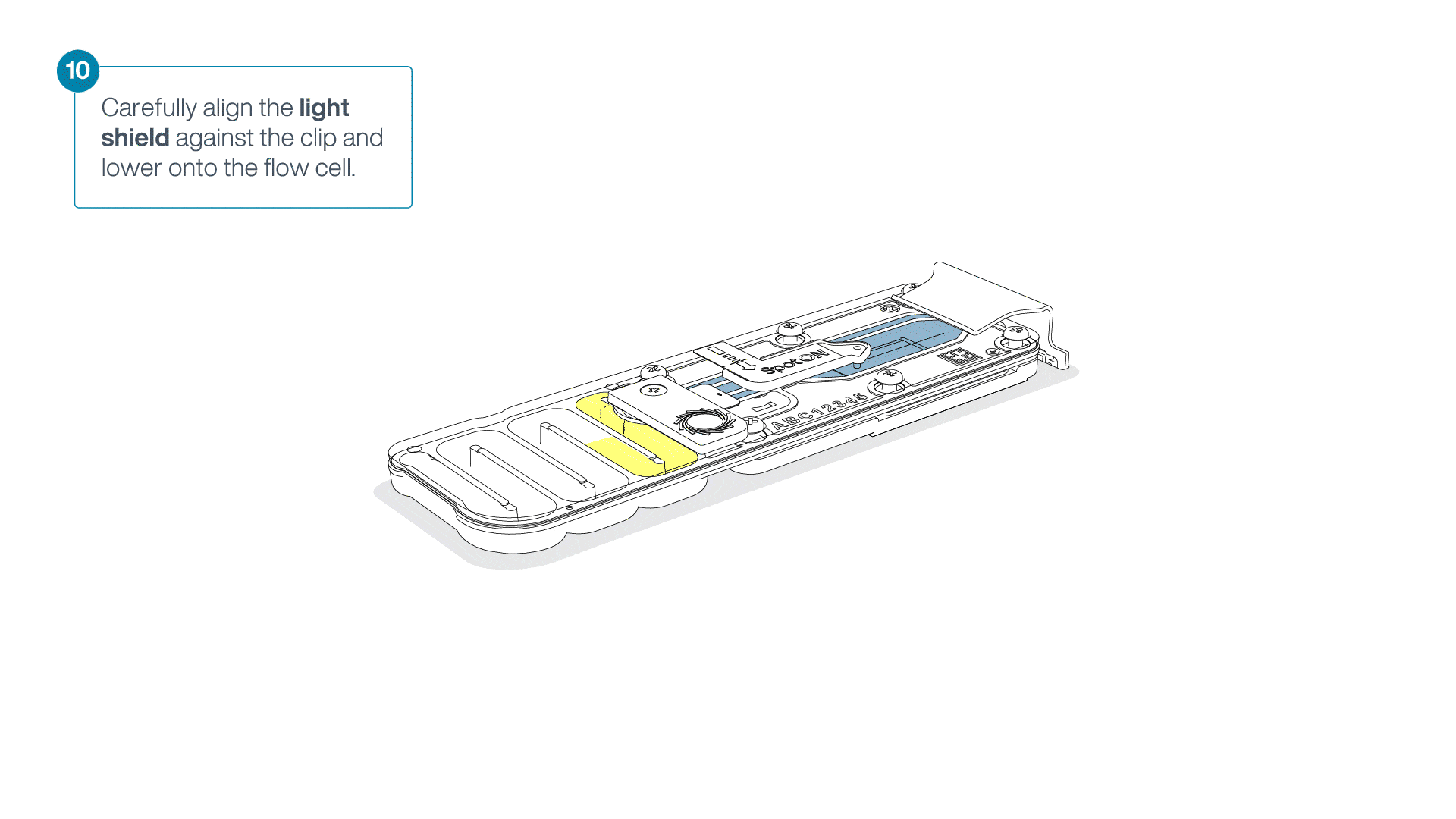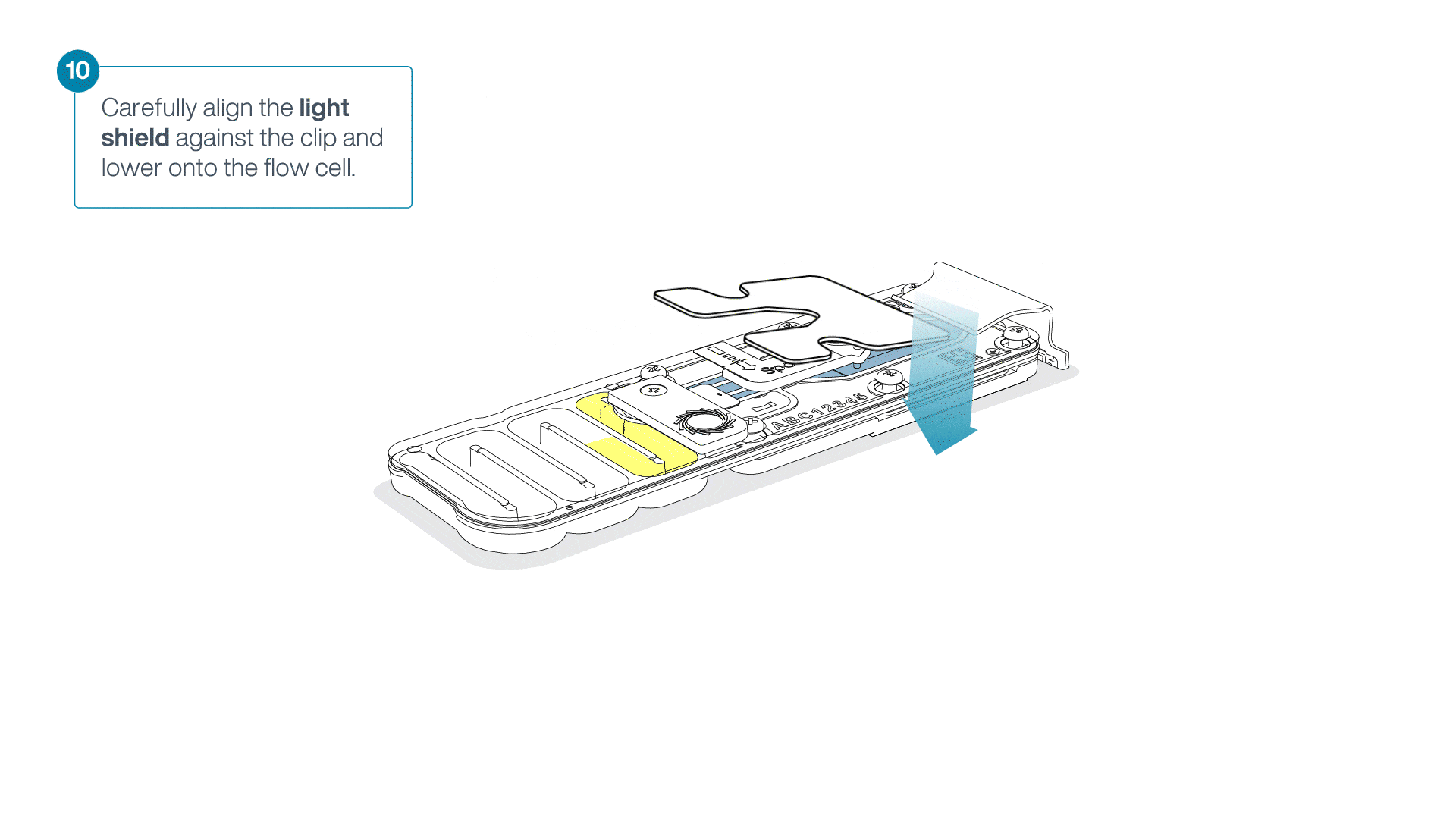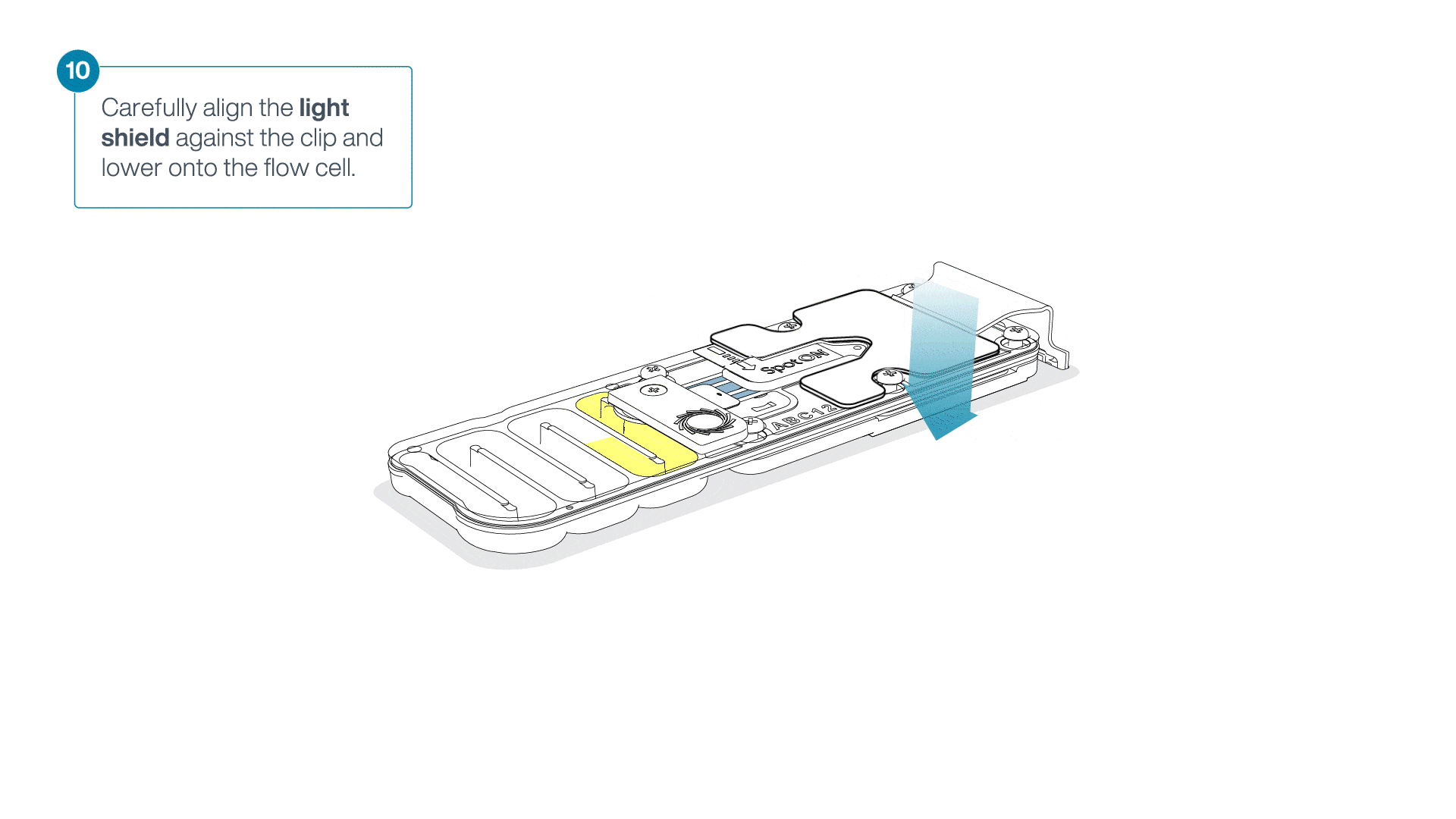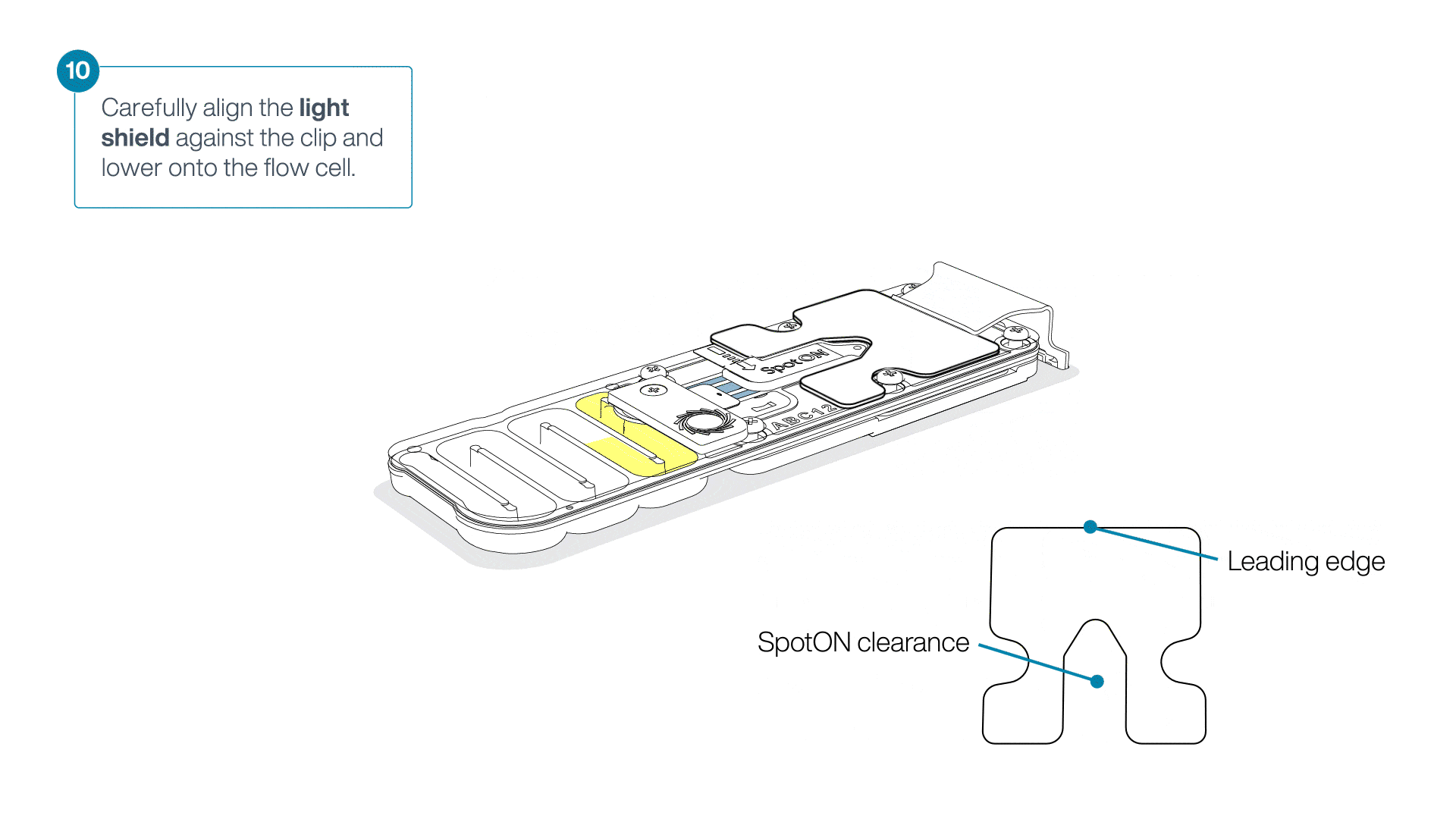
Place the light shield onto the flow cell, as follows:
Carefully place the leading edge of the light shield against the clip.
Note: Do not force the light shield underneath the clip.Gently lower the light shield onto the flow cell. The light shield should sit around the SpotON cover, covering the entire top section of the flow cell.











Sequencing and data analysis
Data acquisition and basecalling
Data acquisition and basecalling
-
How to start sequencing
Once you have loaded your flow cell, the sequencing run can be started on MinKNOW, our sequencing software that controls the device, data acquisition and real-time basecalling. For more detailed information on setting up and using MinKNOW, please see the MinKNOW protocol.
MinKNOW can be used and set up to sequence in multiple ways:
- On a computer either directly or remotely connected to a sequencing device.
- Directly on a GridION or PromethION 24/48 sequencing device.
For more information on using MinKNOW on a sequencing device, please see the device user manuals:
- MinION Mk1B user manual
- MinION Mk1C user manual
- MinION Mk1D user manual
- GridION user manual
To start a sequencing run on MinKNOW:
1. Navigate to the start page and click Start sequencing.
2. Fill in your experiment details, such as name and flow cell position and sample ID.
3. Select the sequencing kit used in the library preparation on the Kit page.
4. Configure the sequencing and output parameters for your sequencing run or keep to the default settings on the Run configuration tab.
Note: If basecalling was turned off when a sequencing run was set up, basecalling can be performed post-run on MinKNOW. For more information, please see the MinKNOW protocol.
5. Click Start to initiate the sequencing run.
-
Duplex basecalling
Kit 14 chemistry has improved duplex basecalling which requires rebasecalling on Dorado after simplex basecalling has been performed on MinKNOW.
For detailed information on setting up your sequencing run, for both simplex and duplex basecalling, please see the Kit 14 sequencing and duplex basecalling info sheet.
Note: When running Dorado, we recommend stopping other basecalling for the best performance by maximising memory available to Dorado. This can be stopped and restarted when Dorado has finished via the GUI on MinKNOW.
-
Data analysis after sequencing
After sequencing has completed on MinKNOW, the flow cell can be reused or returned, as outlined in the Flow cell reuse and returns section.
After sequencing and basecalling, the data can be analysed. For further information about options for basecalling and post-basecalling analysis, please refer to the Data Analysis document.
In the Downstream analysis section, we outline further options for analysing your data.
Flow cell reuse and returns
Flow cell reuse and returns
- Materials
-
- Flow Cell Wash Kit (EXP-WSH004)
-
After your sequencing experiment is complete, if you would like to reuse the flow cell, please follow the Flow Cell Wash Kit protocol and store the washed flow cell at +2°C to +8°C.
The Flow Cell Wash Kit protocol is available on the Nanopore Community.
-
Alternatively, follow the returns procedure to send the flow cell back to Oxford Nanopore.
Instructions for returning flow cells can be found here.
Downstream analysis
Downstream analysis
-
Post-basecalling analysis
There are several options for further analysing your basecalled data:
1. EPI2ME workflows
For in-depth data analysis, Oxford Nanopore Technologies offers a range of bioinformatics tutorials and workflows available in EPI2ME, which are available in the EPI2ME section of the Community. The platform provides a vehicle where workflows deposited in GitHub by our Research and Applications teams can be showcased with descriptive texts, functional bioinformatics code and example data.
2. Research analysis tools
Oxford Nanopore Technologies' Research division has created a number of analysis tools, that are available in the Oxford Nanopore GitHub repository. The tools are aimed at advanced users, and contain instructions for how to install and run the software. They are provided as-is, with minimal support.
3. Community-developed analysis tools
If a data analysis method for your research question is not provided in any of the resources above, please refer to the resource centre and search for bioinformatics tools for your application. Numerous members of the Nanopore Community have developed their own tools and pipelines for analysing nanopore sequencing data, most of which are available on GitHub. Please be aware that these tools are not supported by Oxford Nanopore Technologies, and are not guaranteed to be compatible with the latest chemistry/software configuration.
Troubleshooting
Issues during DNA/RNA extraction and library preparation
Issues during DNA/RNA extraction and library preparation
-
Below is a list of the most commonly encountered issues, with some suggested causes and solutions.
We also have an FAQ section available on the Nanopore Community Support section.
If you have tried our suggested solutions and the issue still persists, please contact Technical Support via email (support@nanoporetech.com) or via LiveChat in the Nanopore Community.
-
Low sample quality
Observation Possible cause Comments and actions Low DNA purity (Nanodrop reading for DNA OD 260/280 is <1.8 and OD 260/230 is <2.0–2.2) The DNA extraction method does not provide the required purity The effects of contaminants are shown in the Contaminants document. Please try an alternative extraction method that does not result in contaminant carryover.
Consider performing an additional SPRI clean-up step.Low RNA integrity (RNA integrity number <9.5 RIN, or the rRNA band is shown as a smear on the gel) The RNA degraded during extraction Try a different RNA extraction method. For more info on RIN, please see the RNA Integrity Number document. Further information can be found in the DNA/RNA Handling page. RNA has a shorter than expected fragment length The RNA degraded during extraction Try a different RNA extraction method. For more info on RIN, please see the RNA Integrity Number document. Further information can be found in the DNA/RNA Handling page.
We recommend working in an RNase-free environment, and to keep your lab equipment RNase-free when working with RNA. -
Low DNA recovery after AMPure bead clean-up
Observation Possible cause Comments and actions Low recovery DNA loss due to a lower than intended AMPure beads-to-sample ratio 1. AMPure beads settle quickly, so ensure they are well resuspended before adding them to the sample.
2. When the AMPure beads-to-sample ratio is lower than 0.4:1, DNA fragments of any size will be lost during the clean-up.Low recovery DNA fragments are shorter than expected The lower the AMPure beads-to-sample ratio, the more stringent the selection against short fragments. Please always determine the input DNA length on an agarose gel (or other gel electrophoresis methods) and then calculate the appropriate amount of AMPure beads to use. 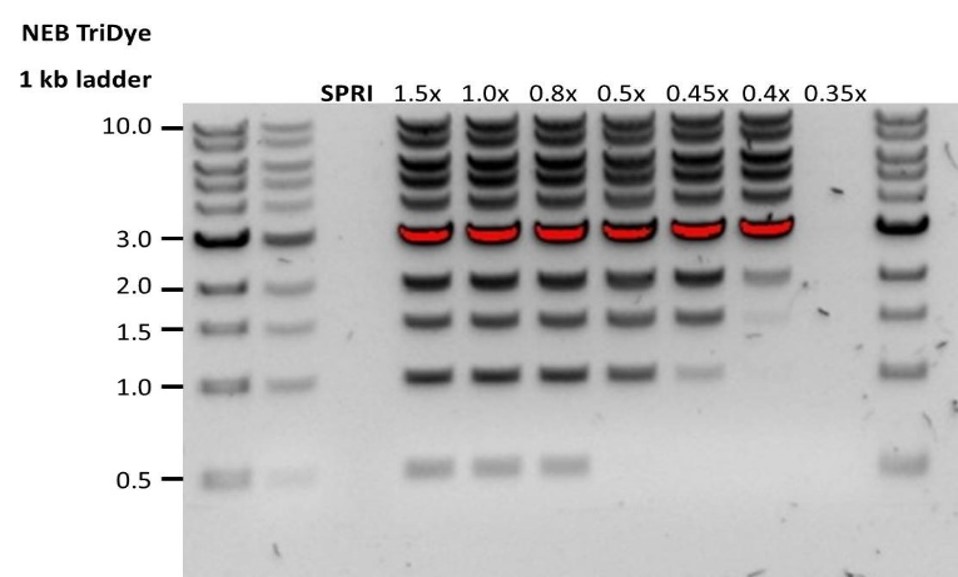
Low recovery after end-prep The wash step used ethanol <70% DNA will be eluted from the beads when using ethanol <70%. Make sure to use the correct percentage.

Issues during the sequencing run
Issues during the sequencing run
-
Below is a list of the most commonly encountered issues, with some suggested causes and solutions.
We also have an FAQ section available on the Nanopore Community Support section.
If you have tried our suggested solutions and the issue still persists, please contact Technical Support via email (support@nanoporetech.com) or via LiveChat in the Nanopore Community.
-
Fewer pores at the start of sequencing than after Flow Cell Check
Observation Possible cause Comments and actions MinKNOW reported a lower number of pores at the start of sequencing than the number reported by the Flow Cell Check An air bubble was introduced into the nanopore array After the Flow Cell Check it is essential to remove any air bubbles near the priming port before priming the flow cell. If not removed, the air bubble can travel to the nanopore array and irreversibly damage the nanopores that have been exposed to air. The best practice to prevent this from happening is demonstrated in this video. MinKNOW reported a lower number of pores at the start of sequencing than the number reported by the Flow Cell Check The flow cell is not correctly inserted into the device Stop the sequencing run, remove the flow cell from the sequencing device and insert it again, checking that the flow cell is firmly seated in the device and that it has reached the target temperature. If applicable, try a different position on the device (GridION/PromethION). MinKNOW reported a lower number of pores at the start of sequencing than the number reported by the Flow Cell Check Contaminations in the library damaged or blocked the pores The pore count during the Flow Cell Check is performed using the QC DNA molecules present in the flow cell storage buffer. At the start of sequencing, the library itself is used to estimate the number of active pores. Because of this, variability of about 10% in the number of pores is expected. A significantly lower pore count reported at the start of sequencing can be due to contaminants in the library that have damaged the membranes or blocked the pores. Alternative DNA/RNA extraction or purification methods may be needed to improve the purity of the input material. The effects of contaminants are shown in the Contaminants Know-how piece. Please try an alternative extraction method that does not result in contaminant carryover. -
MinKNOW script failed
Observation Possible cause Comments and actions MinKNOW shows "Script failed" Restart the computer and then restart MinKNOW. If the issue persists, please collect the MinKNOW log files and contact Technical Support. If you do not have another sequencing device available, we recommend storing the flow cell and the loaded library at 4°C and contact Technical Support for further storage guidance. -
Pore occupancy below 40%
Observation Possible cause Comments and actions Pore occupancy <40% Not enough library was loaded on the flow cell Ensure the correct volume and concentration as stated on the appropriate protocol for your sequencing library is loaded onto the flow cell. Please quantify the library before loading and calculate fmols using tools like the Promega Biomath Calculator, choosing "dsDNA: µg to fmol" Pore occupancy close to 0 The Ligation Sequencing Kit was used, and sequencing adapters did not ligate to the DNA Make sure to use the NEBNext Quick Ligation Module (E6056) and Oxford Nanopore Technologies Ligation Buffer (LNB, provided in the sequencing kit) at the sequencing adapter ligation step, and use the correct amount of each reagent. A Lambda control library can be prepared to test the integrity of the third-party reagents. Pore occupancy close to 0 The Ligation Sequencing Kit was used, and ethanol was used instead of LFB or SFB at the wash step after sequencing adapter ligation Ethanol can denature the motor protein on the sequencing adapters. Make sure the LFB or SFB buffer was used after ligation of sequencing adapters. Pore occupancy close to 0 No tether on the flow cell Tethers are adding during flow cell priming (FLT tube for Kit 9, 10, 11, FCT for Kit 14, and FTU for ultra-long DNA kits). Make sure FLT/FCT/FTU was added to the buffer (FB for Kit 9, 10, 11, and FCF for Kit 14) before priming. -
Shorter than expected read length
Observation Possible cause Comments and actions Shorter than expected read length Unwanted fragmentation of DNA sample Read length reflects input DNA fragment length. Input DNA can be fragmented during extraction and library prep.
1. Please review the Extraction Methods in the Nanopore Community for best practice for extraction.
2. Visualise the input DNA fragment length distribution on an agarose gel before proceeding to the library prep.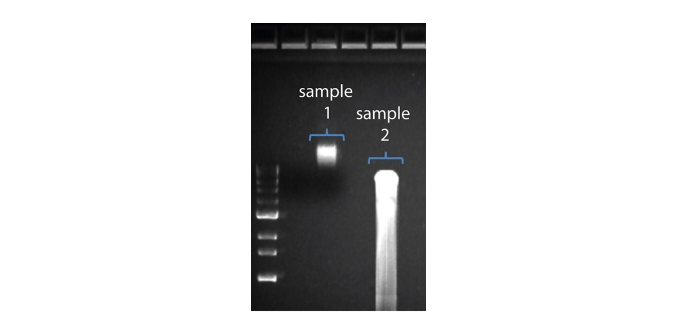 In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented.
In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented.
3. During library prep, avoid pipetting and vortexing when mixing reagents. Flicking or inverting the tube is sufficient. -
Large proportion of unavailable pores
Observation Possible cause Comments and actions Large proportion of unavailable pores (shown as blue in the channels panel and pore activity plot) 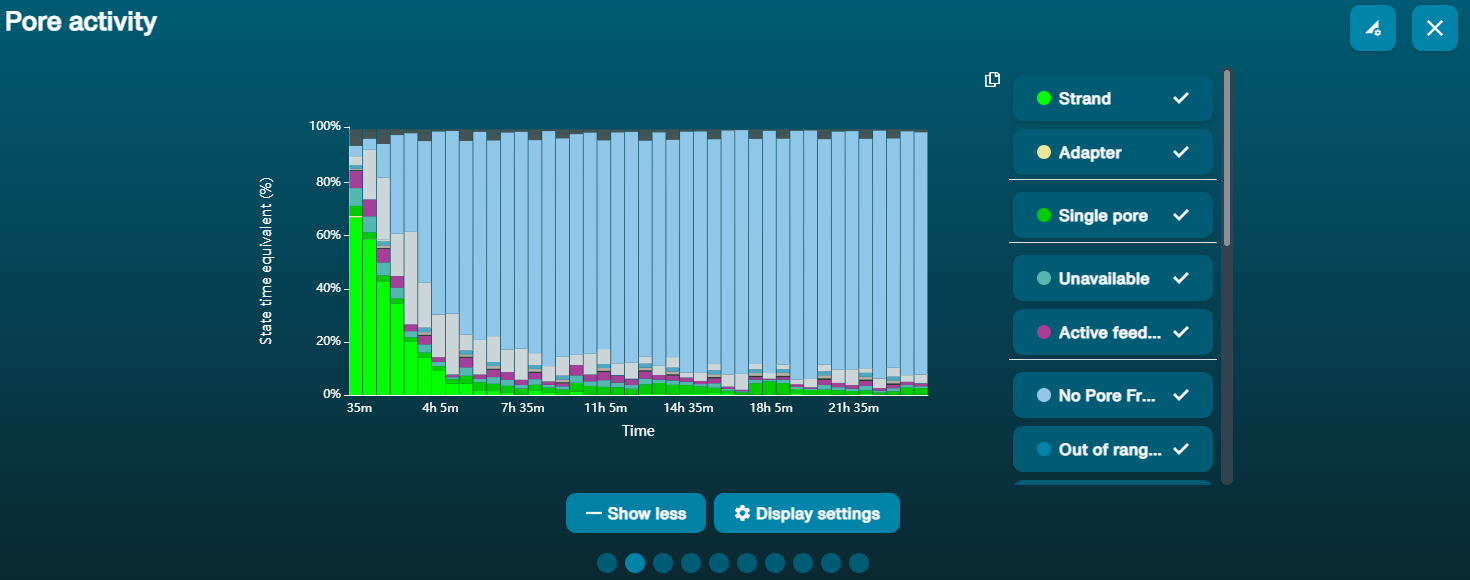 The pore activity plot above shows an increasing proportion of "unavailable" pores over time.
The pore activity plot above shows an increasing proportion of "unavailable" pores over time.Contaminants are present in the sample Some contaminants can be cleared from the pores by the unblocking function built into MinKNOW. If this is successful, the pore status will change to "sequencing pore". If the portion of unavailable pores stays large or increases:
1. A nuclease flush using the Flow Cell Wash Kit (EXP-WSH004) can be performed, or
2. Run several cycles of PCR to try and dilute any contaminants that may be causing problems. -
Large proportion of inactive pores
Observation Possible cause Comments and actions Large proportion of inactive/unavailable pores (shown as light blue in the channels panel and pore activity plot. Pores or membranes are irreversibly damaged) Air bubbles have been introduced into the flow cell Air bubbles introduced through flow cell priming and library loading can irreversibly damage the pores. Watch the Priming and loading your flow cell video for best practice Large proportion of inactive/unavailable pores Certain compounds co-purified with DNA Known compounds, include polysaccharides, typically associate with plant genomic DNA.
1. Please refer to the Plant leaf DNA extraction method.
2. Clean-up using the QIAGEN PowerClean Pro kit.
3. Perform a whole genome amplification with the original gDNA sample using the QIAGEN REPLI-g kit.Large proportion of inactive/unavailable pores Contaminants are present in the sample The effects of contaminants are shown in the Contaminants Know-how piece. Please try an alternative extraction method that does not result in contaminant carryover. -
Temperature fluctuation
Observation Possible cause Comments and actions Temperature fluctuation The flow cell has lost contact with the device Check that there is a heat pad covering the metal plate on the back of the flow cell. Re-insert the flow cell and press it down to make sure the connector pins are firmly in contact with the device. If the problem persists, please contact Technical Services. -
Failed to reach target temperature
Observation Possible cause Comments and actions MinKNOW shows "Failed to reach target temperature" The instrument was placed in a location that is colder than normal room temperature, or a location with poor ventilation (which leads to the flow cells overheating) MinKNOW has a default timeframe for the flow cell to reach the target temperature. Once the timeframe is exceeded, an error message will appear and the sequencing experiment will continue. However, sequencing at an incorrect temperature may lead to a decrease in throughput and lower q-scores. Please adjust the location of the sequencing device to ensure that it is placed at room temperature with good ventilation, then re-start the process in MinKNOW. Please refer to this link for more information on MinION temperature control.
 In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented.
In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented. The pore activity plot above shows an increasing proportion of "unavailable" pores over time.
The pore activity plot above shows an increasing proportion of "unavailable" pores over time.Become a full member
Purchase a MinION Starter Pack from Avantor to get full community access and benefit from:
- News - hear about the latest product updates
- Posts - interact with thousands of nanopore users from around the globe
- Software - download the latest sequencing and analysis software
Already have a Nanopore Community account?
Log in hereNeed more help?
Request a call with our experts for detailed advice on implementing nanopore sequencing.
Request a callInterested in microbiology?
Visit our microbial sequencing spotlight page on vwr.com.
Microbial sequencing