-
Below is a list of the most commonly encountered issues, with some suggested causes and solutions.
We also have an FAQ section available on the Nanopore Community Support section.
If you have tried our suggested solutions and the issue still persists, please contact Technical Support via email (support@nanoporetech.com) or via LiveChat in the Nanopore Community.
-
Fewer pores at the start of sequencing than after Flow Cell Check
Observation Possible cause Comments and actions MinKNOW reported a lower number of pores at the start of sequencing than the number reported by the Flow Cell Check An air bubble was introduced into the nanopore array After the Flow Cell Check it is essential to remove any air bubbles near the priming port before priming the flow cell. If not removed, the air bubble can travel to the nanopore array and irreversibly damage the nanopores that have been exposed to air. The best practice to prevent this from happening is demonstrated in this video. MinKNOW reported a lower number of pores at the start of sequencing than the number reported by the Flow Cell Check The flow cell is not correctly inserted into the device Stop the sequencing run, remove the flow cell from the sequencing device and insert it again, checking that the flow cell is firmly seated in the device and that it has reached the target temperature. If applicable, try a different position on the device (GridION/PromethION). MinKNOW reported a lower number of pores at the start of sequencing than the number reported by the Flow Cell Check Contaminations in the library damaged or blocked the pores The pore count during the Flow Cell Check is performed using the QC DNA molecules present in the flow cell storage buffer. At the start of sequencing, the library itself is used to estimate the number of active pores. Because of this, variability of about 10% in the number of pores is expected. A significantly lower pore count reported at the start of sequencing can be due to contaminants in the library that have damaged the membranes or blocked the pores. Alternative DNA/RNA extraction or purification methods may be needed to improve the purity of the input material. The effects of contaminants are shown in the Contaminants Know-how piece. Please try an alternative extraction method that does not result in contaminant carryover. -
MinKNOW script failed
Observation Possible cause Comments and actions MinKNOW shows "Script failed" Restart the computer and then restart MinKNOW. If the issue persists, please collect the MinKNOW log files and contact Technical Support. If you do not have another sequencing device available, we recommend storing the flow cell and the loaded library at 4°C and contact Technical Support for further storage guidance. -
Pore occupancy below 40%
Observation Possible cause Comments and actions Pore occupancy <40% Not enough library was loaded on the flow cell Ensure you load the recommended amount of good quality library in the relevant library prep protocol onto your flow cell. Please quantify the library before loading and calculate mols using tools like the Promega Biomath Calculator, choosing "dsDNA: µg to pmol" Pore occupancy close to 0 The Ligation Sequencing Kit was used, and sequencing adapters did not ligate to the DNA Make sure to use the NEBNext Quick Ligation Module (E6056) and Oxford Nanopore Technologies Ligation Buffer (LNB, provided in the sequencing kit) at the sequencing adapter ligation step, and use the correct amount of each reagent. A Lambda control library can be prepared to test the integrity of the third-party reagents. Pore occupancy close to 0 The Ligation Sequencing Kit was used, and ethanol was used instead of LFB or SFB at the wash step after sequencing adapter ligation Ethanol can denature the motor protein on the sequencing adapters. Make sure the LFB or SFB buffer was used after ligation of sequencing adapters. Pore occupancy close to 0 No tether on the flow cell Tethers are adding during flow cell priming (FLT/FCT tube). Make sure FLT/FCT was added to FB/FCF before priming. -
Shorter than expected read length
Observation Possible cause Comments and actions Shorter than expected read length Unwanted fragmentation of DNA sample Read length reflects input DNA fragment length. Input DNA can be fragmented during extraction and library prep.
1. Please review the Extraction Methods in the Nanopore Community for best practice for extraction.
2. Visualise the input DNA fragment length distribution on an agarose gel before proceeding to the library prep.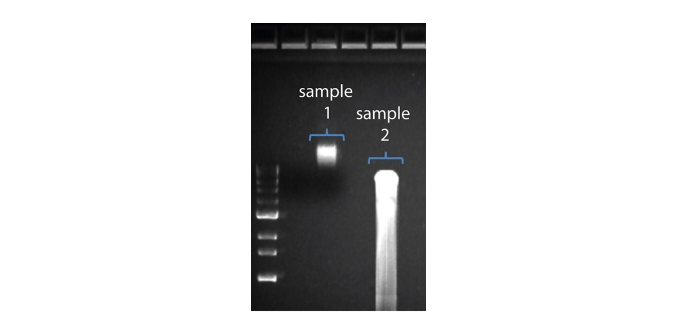 In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented.
In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented.
3. During library prep, avoid pipetting and vortexing when mixing reagents. Flicking or inverting the tube is sufficient. -
Large proportion of unavailable pores
Observation Possible cause Comments and actions Large proportion of unavailable pores (shown as blue in the channels panel and pore activity plot) 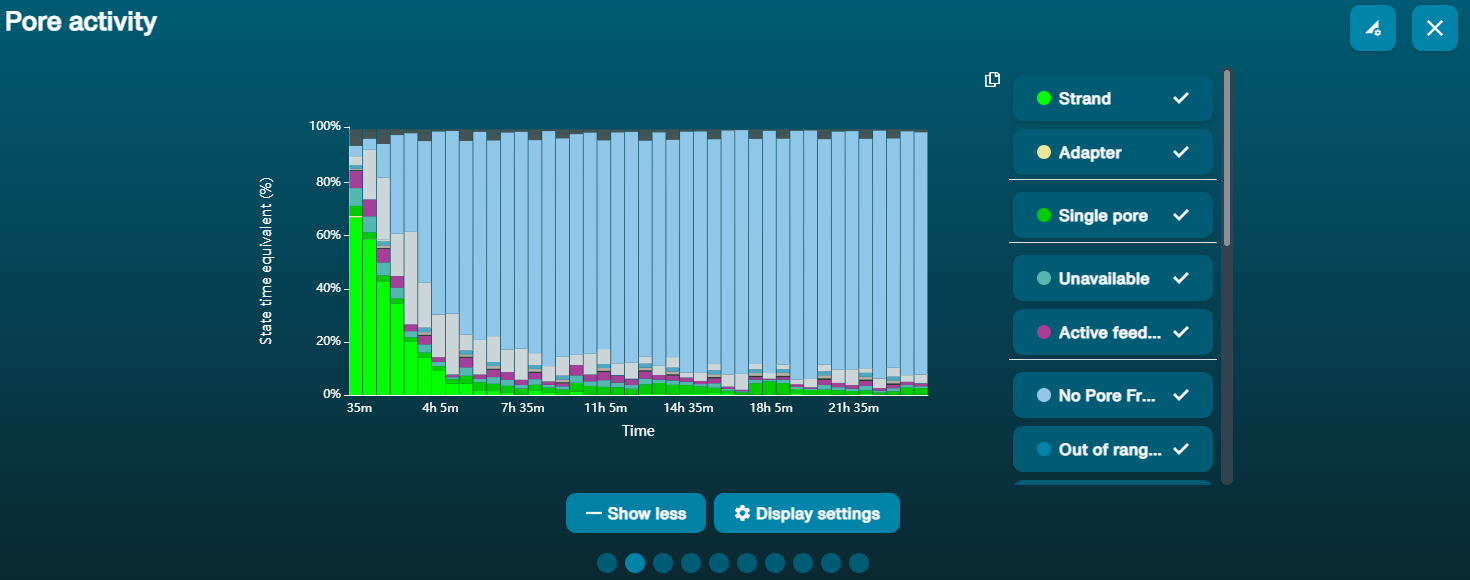 The pore activity plot above shows an increasing proportion of "unavailable" pores over time.
The pore activity plot above shows an increasing proportion of "unavailable" pores over time.Contaminants are present in the sample Some contaminants can be cleared from the pores by the unblocking function built into MinKNOW. If this is successful, the pore status will change to "sequencing pore". If the portion of unavailable pores stays large or increases:
1. A nuclease flush using the Flow Cell Wash Kit (EXP-WSH004) can be performed, or
2. Run several cycles of PCR to try and dilute any contaminants that may be causing problems. -
Large proportion of inactive pores
Observation Possible cause Comments and actions Large proportion of inactive/unavailable pores (shown as light blue in the channels panel and pore activity plot. Pores or membranes are irreversibly damaged) Air bubbles have been introduced into the flow cell Air bubbles introduced through flow cell priming and library loading can irreversibly damage the pores. Watch the Priming and loading your flow cell video for best practice Large proportion of inactive/unavailable pores Certain compounds co-purified with DNA Known compounds, include polysaccharides, typically associate with plant genomic DNA.
1. Please refer to the Plant leaf DNA extraction method.
2. Clean-up using the QIAGEN PowerClean Pro kit.
3. Perform a whole genome amplification with the original gDNA sample using the QIAGEN REPLI-g kit.Large proportion of inactive/unavailable pores Contaminants are present in the sample The effects of contaminants are shown in the Contaminants Know-how piece. Please try an alternative extraction method that does not result in contaminant carryover. -
Reduction in sequencing speed and q-score later into the run
Observation Possible cause Comments and actions Reduction in sequencing speed and q-score later into the run For Kit 9 chemistry (e.g. SQK-LSK109), fast fuel consumption is typically seen when the flow cell is overloaded with library (please see the appropriate protocol for your DNA library to see the recommendation). Add more fuel to the flow cell by following the instructions in the MinKNOW protocol. In future experiments, load lower amounts of library to the flow cell. -
Temperature fluctuation
Observation Possible cause Comments and actions Temperature fluctuation The flow cell has lost contact with the device Check that there is a heat pad covering the metal plate on the back of the flow cell. Re-insert the flow cell and press it down to make sure the connector pins are firmly in contact with the device. If the problem persists, please contact Technical Services. -
Failed to reach target temperature
Observation Possible cause Comments and actions MinKNOW shows "Failed to reach target temperature" The instrument was placed in a location that is colder than normal room temperature, or a location with poor ventilation (which leads to the flow cells overheating) MinKNOW has a default timeframe for the flow cell to reach the target temperature. Once the timeframe is exceeded, an error message will appear and the sequencing experiment will continue. However, sequencing at an incorrect temperature may lead to a decrease in throughput and lower q-scores. Please adjust the location of the sequencing device to ensure that it is placed at room temperature with good ventilation, then re-start the process in MinKNOW. Please refer to this FAQ for more information on MinION Mk 1B temperature control. -
Guppy – no input .fast5 was found or basecalled
Observation Possible cause Comments and actions No input .fast5 was found or basecalled input_path did not point to the .fast5 file location The --input_path has to be followed by the full file path to the .fast5 files to be basecalled, and the location has to be accessible either locally or remotely through SSH. No input .fast5 was found or basecalled The .fast5 files were in a subfolder at the input_path location To allow Guppy to look into subfolders, add the --recursive flag to the command -
Guppy – no Pass or Fail folders were generated after basecalling
Observation Possible cause Comments and actions No Pass or Fail folders were generated after basecalling The --qscore_filtering flag was not included in the command The --qscore_filtering flag enables filtering of reads into Pass and Fail folders inside the output folder, based on their strand q-score. When performing live basecalling in MinKNOW, a q-score of 7 (corresponding to a basecall accuracy of ~80%) is used to separate reads into Pass and Fail folders. -
Guppy – unusually slow processing on a GPU computer
Observation Possible cause Comments and actions Unusually slow processing on a GPU computer The --device flag wasn't included in the command The --device flag specifies a GPU device to use for accelerate basecalling. If not included in the command, GPU will not be used. GPUs are counted from zero. An example is --device cuda:0 cuda:1, when 2 GPUs are specified to use by the Guppy command.
 In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented.
In the image above, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented. The pore activity plot above shows an increasing proportion of "unavailable" pores over time.
The pore activity plot above shows an increasing proportion of "unavailable" pores over time.