-
Optimising your library preparation for duplex data
Duplex basecalling is performed on Dorado after simplex basecalling is completed on MinKNOW. Duplex basecalling is where the complement strand is read immediately after the template strand and the consensus basecall for both strands leads to a further increase in accuracy to ~Q30 with our super-accurate basecaller (SUP).
To generate high duplex output, it is important to follow the library preparation protocol to ensure successful ligation of sequencing adapters onto both ends of the DNA strands. It is also important to follow the flow cell loading recommendation of 10-20 fmols for optimal duplex output to ensure the flow cell is not under- or overloaded:
- Underloading results in lower capture rate and less overall output.
- Overloading adds competition of other strands around the nanopore and reduces the rate of duplex read capture.
For calculating how much DNA to load onto your flow cell, please use the table below for guidance. For more information on DNA input, please see the 'Input DNA/RNA QC' document.Mass Molarity if fragment length = 2 kb Molarity if fragment length = 8 kb Molarity if fragment length = 50 kb 1 μg 770 fmol 193 fmol 31 fmol 500 ng 395 fmol 96 fmol 15 fmol 400 ng 308 fmol 77 fmol 12 fmol 200 ng 154 fmol 39 fmol 6.2 fmol 100 ng 77 fmol 19 fmol 3.1 fmol 75 ng 61 fmol 15 fmol 2.4 fmol 50 ng 40 fmol 10 fmol 1.6 fmol 30 ng 23 fmol 5.8 fmol 0.9 fmol 20 ng 16 fmol 4 fmol 0.6 fmol 15 ng 12 fmol 3 fmol 0.4 fmol 10 ng 7.7 fmol 1.9 fmol 0.3 fmol
To basecall duplex data, we now offer duplex read basecalling in Dorado (recommended) and Guppy, where the template and complement strands of a read can have their basecall data combined to provide a more accurate sequence. For more information, please see our Dorado page on Github.Please note, to generate data of the highest accuracy, ensure data is basecalled using our super-accurate basecaller (SUP).
-
Overview of performing duplex basecalling:
Set up sequencing parameters in MinKNOW to perform simplex basecalling as described in "Basecalling Kit 14 simplex data".
a. Basecall using the Fast basecaller.
b. Output .POD5 files.Using Dorado, re-basecall your simplex data with the following command to output simplex and duplex reads using the super-accurate (SUP) basecalling model. Other basecalling models can be used, as listed on the Dorado Github page.
$ dorado duplex dna_r10.4.1_e8.2_400bps_sup@v4.1.0 pod5s/ > duplex.bamDorado is a high-performance basecaller which is used to perform duplex basecalling. For further information about Dorado, please see the Dorado Github page.
Note: When running Dorado, we recommend stopping other basecalling for the best performance by maximising memory available to Dorado. This can be stopped and restarted when Dorado has finished via the GUI on MinKNOW.
Guppy may also be used to duplex basecall.
-
Calculating duplex data
Duplex data can be presented as two values:
- Duplex rate as a percentage of bases or reads in a duplex pair
- Duplex output as Gb of data in a duplex pair
To determine total percentage of duplex reads, we calculate as follows:= ((template + complement)/total) * 100
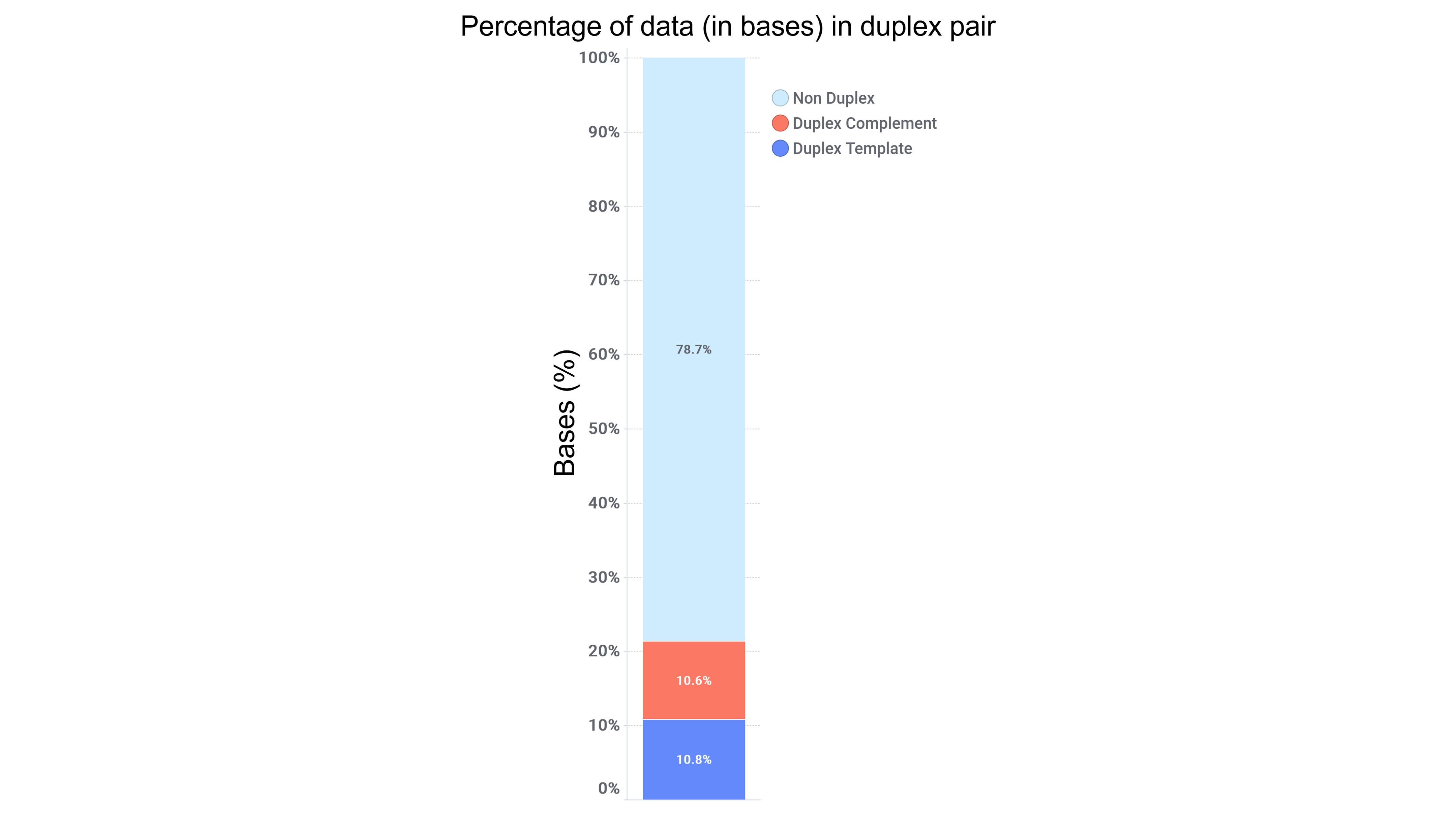
Figure 4. This graph illustrates the duplex reads presented as template strand and complement strand, explaining the need for the calculation to combine the reads to calculate the total percentage. In this example, duplex rate would be 21.4%.
