-
Barcoding/demultiplexing overview
In the Guppy suite, barcoding can be performed by a separate executable. This allows barcoding to be performed as an offline analysis step without having to re-basecall the source reads. To perform barcoding in this way, invoke the barcoder with the minimum required parameters:
guppy_barcoder --input_path <folder containing FASTQ and/or FASTA files> --save_path <output folder> --config configuration.cfg
When performing barcode detection, Guppy will create a
barcoding_summary.txtfile in the output folder, which contains information about the best-matching barcodes for each read in the FASTQ/FASTA files in the input folder (see "Summary file contents" in the "Input and output files" section for details). The output FASTQ/FASTA files will be written into barcode-specific subdirectories for the barcode detected. A log file is also emitted with information about the execution run. -
The Guppy barcoder supports the following optional parameters:
- Version (
-vor--version): Prints the version of Guppy barcoder. - Help (
-hor--help): Print a help message describing usage and all the available parameters.
Data features
- Require a barcode on both ends of the read (
--require_barcode_both_ends): Option to only classify reads where a barcode has been detected at both the front and rear of the read. This can significantly reduce the number of reads that are classified, and is also not a valid argument for the Rapid kits (which do not have a rear barcode). - Allow inferior barcodes to be used in arrangements (
--allow_inferior_barcodes): Option to still classify reads when the barcode selected at each end of the read was not the highest-scoring barcode detected (assuming one was detected above the minimum score). This can slightly increase the number of reads that are classified but can increase the false-positive rate in classifications. - Front window size (
--front_window_size): Specify the maximum window of the start of the read (in bases) to search for the front barcode in. The default is 150 bases. - Rear window size (
--rear_window_size): Specify the maximum window of the end of the read (in bases) to search for the rear barcode in. The default is 150 bases. - Detect mid-strand barcodes (
--detect_mid_strand_barcodes): Flag option to enable detection of barcodes within the strand. This option can be used to detect abnormal reads such as chimeras. If a mid-strand barcode is detected, the read will be classified as "unclassified". - Detect mid-strand adapters (
--detect_mid_strand_adapter): Flag option to enable detection of adapter sequences within the strand. This option can be used to detect abnormal reads such as chimeras. - Minimum score for barcode detection (
--min_score_barcode_front): Specify the minimum score for barcode detection. Unless a minimum score is also set for rear barcodes, this score will be used for both front and rear barcodes. Default is 60. - Minimum score for rear barcodes (
--min_score_barcode_rear): Specify the minimum score for rear barcodes. Use this if you want to set a different minimum score for rear barcodes than for front barcodes. Default is to use the front barcode minimum. - Minimum score for detection of barcode contexts (
--min_score_barcode_mask): Specify the minimum score to consider a barcode context to be a valid location to search for a barcode. If set to -1.0, this option is ignored and barcode scoring is performed on a weighted average of the barcode and context score. Default is -1.0. - Minimum score for detection of mid-strand barcodes (
--min_score_barcode_mid): Minimum score to consider a barcode detected mid-strand to be considered a valid alignment. Mid-strand barcodes below this threshold will be ignored. The default is 40.0. - LamPORE kit (
lamp_kit): Specify the LamPORE kit to use for detection. Note that unlike--barcode_kits, it is not supported to analyse reads against multiple LamPORE kits simultaneously. - Minimum score for detection of LAMP FIP barcodes (
--min_score_lamp): Specify the minimum score to consider a LAMP FIP barcode to be classified. Default is 80.0. - Minimum score for detection of LAMP FIP barcode masks (
--min_score_lamp_mask): Specify the minimum score to consider a LAMP FIP barcode context to be a valid location to search for a FIP barcode. Default is 50.0. - Minimum score for detection of LAMP targets (
--min_score_lamp_target): Specify the minimum score to consider a LAMP target sequence alignment to be classified. Default is 75. - Minimum score for detection of adapters (
--min_score_adapter): Minimum score for an adapter to be considered a valid alignment. Default is 60. - Minimum score for detection of mid-strand adapters (
--min_score_adapter_mid): Minimum score for a mid-strand adapter to be considered a valid alignment. Default is 50. - Minimum score for detection of primers (
--min_score_primer): Minimum score for a primer to be considered to be a valid alignment. Default is 60. - Minimum length for detection of LAMP FIP barcode masks (
--min_length_lamp_context): Specify the minimum length to consider a LAMP FIP barcode context to be a valid location to search for a FIP barcode. Default is 40. - Minimum length for detection of LAMP targets (
--min_length_lamp_target): Specify the minimum length to consider a LAMP target sequence alignment to be classified. Default is 80. - Additional LAMP barcode context bases (
--additional_lamp_context_bases): Number of bases from a lamp FIP barcode context to append to the front and rear of the FIP barcode before performing matching. Default is 2. - Detect adapter sequences at front and rear of the read (
--detect_adapter): Enables adapter detection. Disabled by default. - Detect primer sequences at front and rear of the read (
--detect_primer): Enables primer detection. Disabled by default. - Enable trimming barcodes (
--enable_trim_barcodes): Flag to enable trimming of barcodes from the sequences in the output files. If present, detected barcodes will be trimmed from the sequence. See "Barcode trimming" for more details and related options.
Input/output
- Quiet mode (
-zor--quiet): This option prevents the Guppy basecaller from outputting anything to stdout. Stdout is short for “standard output” and is the default location to which a running program sends its output. For a command line executable, stdout will typically be sent to the terminal window from which the program was run. - Verbose logging (
--verbose_logs): Flag to enable verbose logging (outputting a verbose log file, in addition to the standard log files, which contains detailed information about the application). Off by default. - Recursive (
-ror--recursive): search through all subfolders contained in the--input_pathvalue, and perform barcode detection on any FASTQ or FASTA files found in them. - Configuration file (
-cor--config): This option allows you to specify a configuration file, which contains details of the parameters used during barcode detection. The default configuration file supplied with Guppy should be sufficient for most users. There is an additional configuration_dual.cfg containing settings for using dual-barcode preparations. - Override default data path (
-dor--data_path): Option to explicitly specify the path to use for loading any data files the application requires (for example, if you have created your own model files or config files). - Records per FASTQ (
-qor--records_per_fastq): The maximum number of reads to put in a single FASTQ or FASTA file. Set this to zero to output all reads into one file (per run id, per batch). The default value is 4000. - Perform FASTQ compression (
-–compress_fastq): Flag to enable gzip compression of output FASTQ/FASTA files; this reduces file size to about 50% of the original. See also--read_batch_size. - BAM file output (
--bam_out): This flag enables BAM file output. Default is for BAM file output to be disabled. - BAM file indexing (
--index): This flag enables BAM file indexing. If the flag is present,guppy_barcodersorts the BAM file output and generates the BAI index file. This flag requires that--bam_outis also set. Disabled by default. - FASTQ file output (
--fastq_out): This flag enables FASTQ file output. If neither--bam_outor--fastq_outis enabled, FASTQ output is enabled by default. - Input valid extensions (
--ext_in): Only files with the specified extensions are processed (comma separated list). If this is not enabled, all files with supported extension are processed. Supported extensions are:.fastq,.fq,.fasta,.fa,.sam,.bam. Sequences from a.samor.bamfile that have been stored as the reverse complement will be reverse-complemented before barcoding.
Optimisation
- Worker thread count (
-tor--worker_threads): The number of worker threads to spawn for the barcoder to use. Increasing this number will allow Guppy barcoder to make better use of multi-core CPU systems, but may impact overall system performance. - GPU device (
-xor--device): Specify the CUDA-enabled GPU to use to perform barcode alignment. Parameters are specified the same way as in the basecaller application. - Limit the kits to detect against (
--barcode_kits): List of barcoding kit(s) or expansion kit(s) used to limit the number of barcodes to be detected against. This speeds up barcoding. Multiple kits must be a space-separated list in double quotes. - Number of parallel GPU barcoding buffers (
--num_barcoding_buffers): Number of parallel memory buffers to supply to the GPU for barcode strand detection. Greater numbers will increase parallelism on the GPU at an increased memory cost. The default is 24. - Number of reads to process in parallel in each GPU barcoding buffer (
--num_reads_per_barcoding_buffer): The number of reads to process in parallel in each GPU barcoding buffer. Greater numbers will increase parallelism on the GPU at an increased memory cost. The default is 4. - Number of parallel GPU mid-barcode detection buffers (
--num_mid_barcoding_buffers): Number of parallel memory buffers to supply to the GPU for barcode mid-strand detection. Greater numbers will increase parallelism on the GPU at an increased memory cost. The default is 96. - Limit the barcodes to a subset of the kits (
--barcode_list): Only the barcodes in this space-separated list will be considered when barcoding. - Progress stats reporting frequency (
--progress_stats_frequency): Frequency in seconds in which to report progress statistics, if supplied will replace the default progress display. - Trace catagory logs (
--trace_category_logs): Enable trace logs - list of strings with the desired names. - Trace domains config (
--trace_domains_config): Configuration file containing list of trace domains to include in verbose logging (if enabled) - Disable pings (
--disable_pings): Flag to disable sending any telemetry information to Oxford Nanopore Technologies. See the "Ping information" section for a summary of what is included in the Guppy telemetry. - Telemetry URL (
--ping_url): Override the default URL for sending telemetry pings. - Ping segment duration (
--ping_segment_duration): Duration in minutes of each ping segment.
To see the supported barcoding kits, run the
--print_kitsargument with the barcoder:
guppy_barcoder --print_kits
To limit the kits to detect against:
guppy_barcoder --input_path <folder containing FASTQ and/or FASTA files> --save_path <output folder> --config configuration.cfg --barcode_kits SQK-RPB004
Or for multiple kits add a space-separated list in double quotes:
guppy_barcoder --input_path <folder containing FASTQ and/or FASTA files> --save_path <output folder> --config configuration.cfg --barcode_kits "EXP-NBD104 EXP-NBD114"
Barcoding of dual-barcode arrangements is also supported. To use dual-barcode arrangements, the correct configuration file must be specified:
guppy_barcoder --input_path <folder containing FASTQ and/or FASTA files> --save_path <output folder> --config configuration_dual.cfg --barcode_kits "EXP-DUAL00"
Note that running barcode detection on dual- and single- barcode kits at the same time is not currently supported. New columns will be emitted into the
barcoding_summary.txtorsequencing_summary.txtwhen performing demultiplexing of dual barcode kits:barcode_front_id_inner,barcode_front_score_inner,barcode_rear_id_innerandbarcode_rear_score_inner. - Version (
-
Barcoding during basecalling
It is also possible to perform barcode detection during the basecalling process. When invoking the
guppy_basecallerexecutable, simply provide a valid set of kits to thebarcode_kitsargument to enable barcoding, for example:
guppy_basecaller --input_path <folder containing .fast5 or .pod5 files> --save_path <output folder> --config dna_r9.4.1_450bps_fast.cfg --barcode_kits SQK-RBK001
Note that options such as barcode trimming and demultiplexing output FASTQ/FASTA files are all supported by the
guppy_basecallerexecutable as well asguppy_barcoder. Guppy also supports barcoding demultiplexing during basecalling when using theguppy_basecall_server. If a barcoding configuration file other than the default configuration.cfg is required, the basecaller executable supports selecting a barcode config using--barcoding_config_filecommand-line option. -
Barcode FASTQ output
The barcoding executable will output FASTQ/FASTA files into barcode-specific subdirectories in the output folder depending on the barcode that was detected. The FASTQ naming follows the same rules as for basecalling (see "Guppy features, settings and analysis"). A barcode directory will only exist if the barcode was detected. The output structure will look like this:
guppy_output_folder/
| barcoding_summary.txt
--- barcode01/
| fastq_runid_777_0.fastq
| fastq_runid_abc_0.fastq
| fastq_runid_abc_1.fastq
--- barcode03/
| fastq_runid_777_0.fastq
| fasta_runid_xyz_0.fasta
--- unclassified/
| fastq_runid_777_0.fastq
-
Barcode trimming
The barcoding executable can automatically trim the detected barcodes from the sequence before being output to the FASTQ/FASTA file. This is off by default. To enable barcode trimming add the
--enable_trim_barcodesargument:
guppy_barcoder --input_path <folder containing FASTQ and/or FASTA files> --save_path <output folder> --config configuration.cfg --enable_trim_barcodes
Two extra columns will then be written into the
barcoding_summary.txtoutput:barcode_front_total_trimmedandbarcode_rear_total_trimmed. A barcode will only be trimmed if it is above themin_scorethreshold (default 60), and the aligned sequence that matches to the barcode will be removed from the front and/or rear of the sequence that is then written to the FASTQ/FASTA.If the user wants to be more severe with trimming, there is a
--num_extra_bases_trimargument, which defaults to 0. Setting this to, for example, 2 would trim the detected barcode sequence plus an extra 2 bases. If the user wants to be more cautious then give this argument a negative number; for example, -3 would trim 3 fewer bases than was detected as the barcode sequence.
guppy_barcoder --input_path <folder containing FASTQ and/or FASTA files> --save_path <output folder> --config configuration.cfg --num_extra_bases_trim 2
-
Expert users - adjusting barcode classification thresholds
The classification threshold has been chosen to produce a low number of incorrect classifications while retaining an acceptable classification rate. The user may override this, but note that small changes can have a significant effect on the false-positive rate, so it is important to always test any changes before using them.
To change the threshold used for both the front and rear barcode modify the
--min_scoreargument. The following would increase the threshold for barcodes to be classified to 70, so that if either the front or rear barcode has a score of 70 or more the read will be classified:
guppy_barcoder --input_path <folder containing FASTQ and/or FASTA files> --save_path <output folder> --config configuration.cfg --min_score 70
The user may also have different front and rear thresholds by also supplying the
--min_score_rear_overrideargument. If this is specified then--min_scorewill be used for the front barcode and--min_score_rear_overridewill be used for the rear barcode. For example, in the following a read will be classified if either the front barcode is above the default (which is currently 60), or the rear barcode 55 or more:
guppy_barcoder --input_path <folder containing FASTQ and/or FASTA files> --save_path <output folder> --config configuration.cfg --min_score_rear_override 55
-
How barcode demultiplexing works in Guppy
This is a general outline of how the Guppy barcoder works and how you can adjust its classification thresholds.
-
The regions of a barcode
A complete barcode arrangement comprises three sections:
- The upstream flanking region, which comes between the barcode and the sequencing adapter
- The barcode sequence
- The downstream flanking region, which comes between the barcode and the sample sequence
A complete dual-barcode arrangement comprises five sections:
- The upstream flanking region, which comes between the outer barcode and the sequencing adapter
- The outer barcode sequence
- The mid flanking region, which comes between the outer barcode and the inner barcode
- The inner barcode sequence
- The downstream flanking region, which comes between the inner barcode and the sample sequence
The barcode sequences remain constant across almost all of Oxford Nanopore Technologies' kits. For example, the flanking regions for barcode 10 in the Rapid Barcoding Kit (SQK-RBK004) are different from the flanking regions for barcode 10 in the native barcoding expansion kit (EXP-NBD114), but the barcode sequence itself is the same.
While native kits use the same barcode sequences as other kits, barcodes 1-12 in the native kit are the reverse complement of the standard barcodes 1-12.
There is one other exception to this: barcode 12a in the Rapid PCR Barcoding Kit SQK-RPB004 has a different barcode sequence to barcode 12 in other kits. For this reason, the oligonucleotide of this sequence is referred to as "barcode 12a".
-
Different barcoding chemistries
While each barcoding chemistry type (e.g. native, rapid, or PCR) will produce barcodes with the pattern described in "The regions of a barcode", there can be variations in the flanking regions within a particular kit. These are referred to as either "forward" and "reverse" variations or "variation 1" and "variation 2" depending on the configuration. When these variations are present the full double-stranded sequence can look like this:
<barcodeXX_var1---><sample sequence top strand---><barcodeXX_var2_rc><barcodeXX_var1_rc><sample sequence bottom strand><barcodeXX_var2--->The PCR Barcoding Expansion kit (EXP-PBC001) produces barcodes like the example directly above.
Or like this:
<barcodeXX_var1><sample sequence top strand---><barcodeXX_var2><barcodeXX_var2><sample sequence bottom strand><barcodeXX_var1>The Native Barcoding Expansion kit (EXP-NBD114) produces barcodes like the second example, directly above.
-
The barcoding algorithm
The barcoding algorithm uses a modified Needleman-Wunsch method. We modify the Needleman-Wunsh algorithm by adding "gap open" and "gap extension" penalties, as well as separate "start gap" and "end gap" penalties. These penalties and the match / mismatch scores for aligning a barcode to a sequence are detailed in two places:
Generic gap penalties are in the barcoding configuration file configuration.cfg, or configuration_dual.cfg for dual-barcode arrangements.
DNA-specific match/mismatch scores are stored in the file
4x4_mismatch_matrix.txt. Note that these scores are shifted such that the highest score is 100 – this means that the final barcode score will share the same maximum. There is also a5x5_mismatch_matrix.txtfile which includes the ability to match any cardinal base to a mask base 'N'.Each barcode is aligned to a section of the basecall, usually the first and / or last 150 bases. This generates a grid of size 150 * < barcode_length >.
The barcoding score for a particular grid is calculated in a two-step process:
The score for only the section of the grid that corresponds to the barcode itself is considered. This corresponds to removing the initial gap row and discarding all scores past the alignment of the last base of the barcode, or removing those sections where the "start gap" and "end gap" penalties are applied.
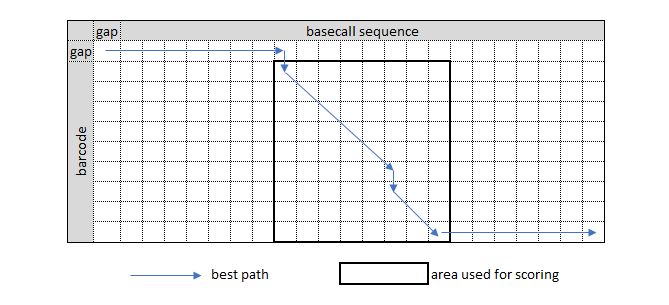
The score is normalized by the total length of the barcode sequence. This ensures the final score is no more than the highest score in the mismatch table (which should be 100). Note that this potentially allows for negative scores when there are a relatively high number of gaps and/or mismatches.
-
Measuring classification
The classification for a particular barcode is determined by comparing the barcoding score to a fixed classification threshold – scores that exceed the threshold are considered (successful) classifications. The current threshold is set to 60 for single barcode arrangements and 50 for dual barcode arrangements.
Classification for a read is determined by taking the single highest-scoring (successful) barcode classification. This includes both classifications made at the beginning of the sequence and (where applicable) the end. If no classification exists then the read is considered "unclassified".
-
Classification threshold criteria
The classification threshold has been chosen to produce a low number of incorrect classifications while retaining an acceptable classification rate. This means that when a read has been classified as having a particular barcode, that classification will be incorrect a low number of times. Ideally this false-positive rate is around 1 in 1000, though this can be dependent on how well individually-barcoded samples are purified before they are pooled together. Classification rates should be 90% or above for samples with barcodes on both ends.
It is important to note that the above evaluation criteria assume that only reads which pass Guppy's quality filters are used. This corresponds to reads which are placed in the "pass" folder after basecalling; generally these will be reads with a mean q-score value greater than 7.
-
Modifying classification thresholds
It is possible to increase the number of classifications at the cost of the false-positive rate. Small changes to this can have a significant effect on the false-positive rate, so it is important to test any changes to the thresholds before using them.
For example, here is a graph of the number of reads classified for particular binned values of the (best) barcoding score. The data set is a collection of around 200,000 reads barcoded with the Native Expansion kit (EXP-NBD114):
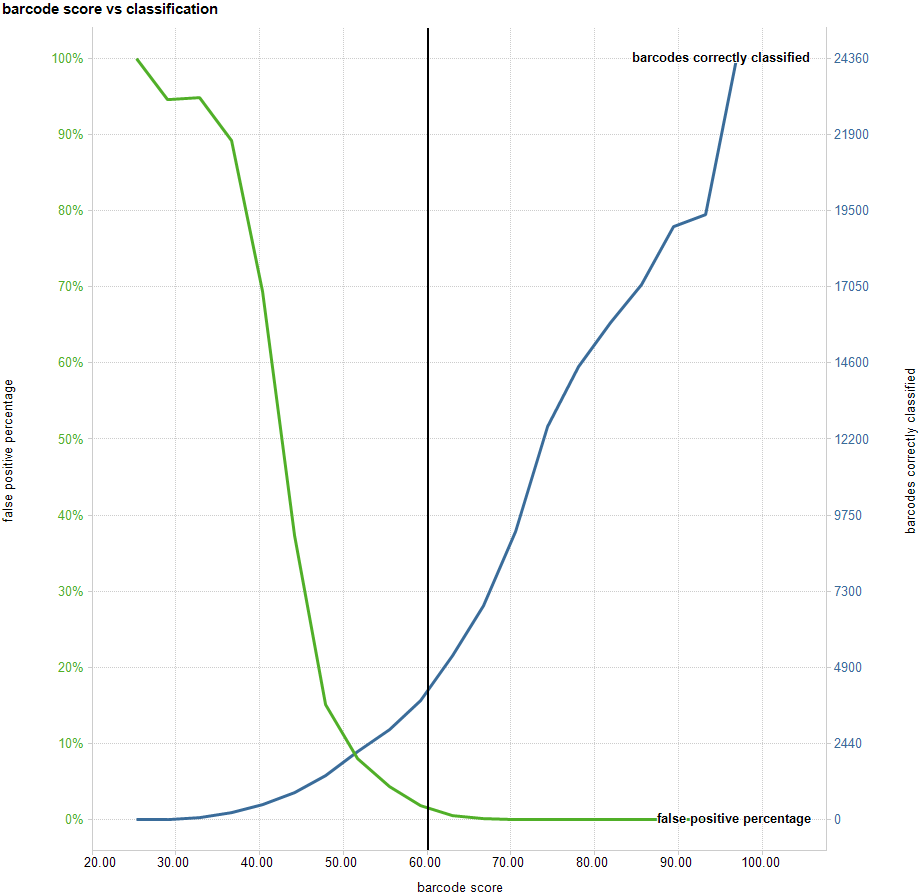
This graph shows that, for example, reads where the best barcode score is around 30 will have about ~95% incorrect classifications. In contrast, for those reads where the highest barcode score is around 95 there will be near 0% incorrect classifications, and we correctly classify around 22,000 reads.
By reducing the threshold by a few points additional correct classifications may be obtained, but the cost in false positive percentage can go up significantly.
The threshold may be changed by modifying the
--min_scoreargument, which applies the threshold to both the front and rear barcode. To have different thresholds for the front and rear barcode modify the--min_score_rear_overrideargument to change the rear barcode threshold. In that case the--min_scoreargument will apply to only the front barcode. -
How classifications are reported
When barcodes are loaded into Guppy for classification, they are loaded in arrangements. An arrangement consists of either:
- One barcode, when searching for barcodes only at the front of a read.
- A front barcode and a rear barcode, when searching for barcodes at both ends of a read.
Once the classification for a particular read has been determined (by choosing the single highest-scoring barcode alignment), there may be another barcode in the arrangement corresponding to the other end of the read. The score for this barcode is also retrieved and reported, regardless of its classification – this means the entire arrangement is always reported.
For example, if a barcode arrangement is loaded containing
barcode01_FWD + barcode01_REVwithbarcode01_FWDmatching the front of the read with a score of 90 andbarcode01_REVmatching the rear of the read with a score of 10, then the final reported result will be:
front_barcode: barcode01_FWD
front_score: 90
rear_barcode: barcode01_REV
rear_score: 10
-
Adding your own barcodes
Guppy fully supports the use of custom barcode sequences. It is recommended that this is accomplished by copying Guppy's existing configuration files and modifying them elsewhere with a text editor.
Barcoding data files
Barcoding data files are contained in Guppy's data folder in the "barcoding" subfolder. You can find this folder in the following locations:
On Linux:
- In
/opt/ont/guppy/dataif installing from deb or RPM. - In the
datafolder in the main Guppy directory if installing from archive.
On OS X/macOS:
- In the
datafolder in the main Guppy directory.
On Windows:
- In
C:\Program Files\Oxford Nanopore\ont-guppy-cpu\data
This folder contains the following subfolders and types of files:
4x4_mismatch_matrix.txt the DNA mismatch matrix for aligning barcodes to sequences
5x5_mismatch_matrix.txt the DNA mismatch matrix for aligning barcodes to sequences including a
'N' mask base
5x5_mismatch_matrix_simple.txt the DNA mismatch matrix for use with dual barcodes.
barcodes_masked.fasta the full list of all barcode and the flanking region mask sequences
lamp_targets.fasta the full list of all LamPORE kit target sequences
configuration.cfg the configuration file containing parameters used in barcode detection
barcoding_arrangements/
barcode_arrs_XXX.toml the arrangement files for specific barcodes
barcoding_dual_arrangements/
barcode_arrs_dual_XXX.toml the arrangement files for specific dual barcodes
lamp_arrangements/
barcode_arrs_lampXXX.toml the arrangement files for specific LamPORE kit configurations
4x4 mismatch_matrix.txt: A tab-delimited file containing the mismatch penalties for DNA.5x5 mismatch_matrix.txt: A tab-delimited file containing the mismatch penalties for DNA plus a masking base 'N', which matches against all bases with a score of 90.5x5 mismatch_matrix_simple.txt: A tab-delimited file containing the mismatch penalties for DNA plus a masking base 'N', which matches against all bases with a score of 90. This version of the 5x5 mismatch matrix has been optimised for dual-barcoding arrangements.barcoding_arrangementsfolder: Folder containing barcoding arrangement files.barcoding_dual_arrangementsfolder: Folder containing dual barcoding arrangement files.lamp_arrangementsfolder: Folder containing arrangement files for LamPORE kits.example_barcode_arrs_XXX.toml(andexample_barcode_arrs_dual_XXX.toml): A .toml formatted arrangement file describing how a particular set of barcode arrangements is configured. It contains the following fields:
[loading options]
barcodes_filename = [filename]
double_variants_frontrear = [true / false]
[arrangement]
name = [name of the barcoding arrangement]
id_pattern = [barcode id pattern]
compatible_kits = [array of kits]
first_index = [first barcode number to load]
last_index = [last barcode number to load]
kit = [kit name]
normalised_id_pattern = ["barcode_arrangement" summary file pattern]
scoring_function = "MAX"
barcode1_pattern = [pattern to look up front barcode in [barcodes_filename]]
barcode2_pattern = [pattern to look up rear barcode in [barcodes_filename]]
mask1 = [mask name to look up front barcode masking region in [barcodes_filename] (optional)]
mask2 = [mask name to look up rear barcode masking region in [barcodes_filename] (optional)]
barcode_inner1_pattern = [pattern to look up front inner barcode in [barcodes_filename] when dual barcoding (optional)]
barcode_inner2_pattern = [pattern to look up rear inner barcode in [barcodes_filename] when dual barcoding (optional)]
barcode_arrs_lampXXX.toml: A .toml formatted arrangement file describing how a LamPORE arrangement is configured. These are similar to barcoding arrangement files, but support a slightly different set of:
[loading options]
barcodes_filename = [filename]
lamp_targets_filename = [filename containing sequences which should be used as LAMP targets]
double_variants_frontrear = [true / false]
[arrangement]
name = [name of the lamp arrangement]
id_pattern = [barcode id pattern]
compatible_kits = [array of kits]
first_index = [first barcode number to load]
last_index = [last barcode number to load]
kit = [kit name]
normalised_id_pattern = ["barcode_arrangement" summary file pattern]
scoring_function = "MAX"
barcode1_pattern = [pattern to look up front barcode in [barcodes_filename]]
lamp_masks = [An array of mask names look up barcode masking regions in [barcodes_filename]]
These sections are dealt with in reverse order.arrangement
id_pattern: The barcode ID pattern itself is what is used as the base name for each barcode arrangement. It may be modified later depending on what is present in theloading_optionssection (see below). This pattern should have%0Nipresent somewhere in the name (whereNis the number of digits to use in the barcode number), as that will be replaced with the barcode number for the arrangement. For example, the patternNB%03iwill be formatted to produce barcode arrangement names such asNB001,NB384, etc. The final arrangement name based on this pattern will be reported in thebarcode_full_arrangementfield in thebarcoding_summary.txtfile.compatible_kits: A list of kits this set of arrangements is compatible with. These may be selected from the command line to restrict the arrangements that barcodes are matched against.first_index: The first integer used when loading barcodes frombarcodes_filename(seeloading optionsbelow). These integers are used to populate the%0Niparts of the barcode name,normalised_id,barcode1, andbarcode2patterns.last_index: The last index used (inclusively) when loading barcodes frombarcodes_filename.kit: The name reported in the "kit" column of the barcoding summary file.normalised_id_pattern: The name reported in the "barcoding_arrangement" column of the barcoding summary file. This should contain the%0Nipattern within it so that the barcode number can be added. This is normally used to report the barcode number without the kit designation.scoring_function: The function used to score a barcode arrangement. There are two choices for this, though only "MAX" is currently used:MAX: The barcode arrangement score is the larger of the front and rear scores.ADD: The barcode arrangement score is the sum of the front and rear scores.
barcode1_pattern: Optional pattern used to look up the front barcode sequences inbarcodes_filename. If this field is not present then no front barcodes will be added during the initial barcode loading step (although it is still possible to obtain front barcodes depending on loading options below). Note that the suffix_FWDwill be added to this barcode name in the arrangement.barcode2_pattern: Optional pattern used to look up the rear barcode sequences inbarcodes_filename. Note that the suffix_REVwill be added to this barcode name in the arrangement, and the barcode inserted into the arrangement will be the reverse complement of the named barcode specified inbarcodes_filename.mask1: Optional. Used to look up the front barcode flanking region inbarcodes_filename. If this field is used, this masking region will be aligned first, then the barcode1 sequence for each arrangement will be aligned to the section of the read which corresponds to the masked-off region of this sequence (i.e. the section of 'N' bases).mask2: Optional. Used to look up the rear barcode flanking region inbarcodes_filename. If this field is used, this masking region will be aligned first, then thebarcode2sequence for each arrangement will be aligned to the section of the read which corresponds to the masked-off region of this sequence (i.e. the section of 'N' bases).barcode_inner1_pattern: Optional pattern used to look up the front inner barcode sequences inbarcodes_filename. Note that the suffix_FWDwill be added to this barcode name in the arrangement.barcode_inner2_pattern: Optional pattern used to look up the rear inner barcode sequences inbarcodes_filename. Note that the suffix_REVwill be added to this barcode name in the arrangement, and the barcode inserted into the arrangement will be the reverse complement of the named barcode specified inbarcodes_filename.lamp_masks: A comma-seperated list of patterns to look up barcode masking regions inbarcodes_filename. Note that there can be several of these masks, as they may be different for each target. The mask will be used to find a context in the sequence to inspect for FIP barcodes.
loading options
barcodes_filename: the name of the FASTA file to load barcodes from. It should be in the data/barcoding folder, or the filename should include a relative path from the data/barcoding folder.lamp_targets_filename: the name of the FASTA file to load LamPORE targets from. It should be in the data/barcoding folder, or the filename should include a relative path from the data/barcoding folder. The targets should be named[target_id]:[specific_sequence_id]. This allows multiple sequences to map to the same target ID. Just thetarget_idwill be reported by the detector.double_variants_frontrear: For each barcode arrangement, create_var1and_var2variants. The_var1variant will bebarcode1at the front andbarcode2at the rear, and_var2will bebarcode2at the front andbarcode1at the rear. This effectively adds a complement for each arrangement.
For example:
loading options barcode names expected in barcodes_filename (assuming both barcode1 and barcode2 patterns are present) barcode arrangements added to list to test against ("rc" denotes reverse complement)
[front_barcode] + [rear_barcode]double_variants_frontrear : false [barcode1]
[barcode2][barcode1]FWD + [barcode2rc]REV double_variants_frontrear : true [barcode1]
[barcode2][barcode1]var1_FWD + [barcode2rc]_var2_REV [barcode2]var2_FWD + [barcode1rc]var1REV Quick start
Assume we have created a set of custom barcodes structured like this:
[barcodeXX---][sample sequence top strand---][barcodeXX_rc]
[barcodeXX_rc][sample sequence bottom strand][barcodeXX---]
There is one type of barcode, it can be attached on both the top and bottom strand, and it has a reverse complement present on the opposite strand.Furthermore, assume we have two different barcodes to add. We will call these two barcodes CUST01 and CUST02.
Step 1: Copy the Guppy data folder to a different location
See "Barcoding data files" above. Assuming a Linux deb installation, this could look something like this:
cp -r /opt/ont/guppy/data ~/mydata
Step 2: Create a new arrangements file and a new FASTA file to store our custom barcodes.
Copy one of the arrangements in the barcoding data folder to store the new arrangement:
cp ~/mydata/barcoding/barcoding_arrangements/barcode_arrs_nb24.toml ~/mydata/barcoding/barcoding_arrangements/barcode_arrs_cust2.toml
And create a new FASTA file for the custom barcodes:
touch ~/mydata/barcoding/custom_barcodes.fasta
Step 3: Edit the new arrangement file to include information on the new barcode
We have one type of barcode, and arrangements of that barcode will include the barcode at the front and the reverse complement of the barcode at the rear. We do not want to setdouble_variants_frontrearbecause we have only one variant of our barcode.The configuration file
barcode_arrs_cust2.tomlwill look like this:
[loading_options]
barcodes_filename = "custom_barcodes.fasta"
double_variants_frontrear = false
[arrangement]
name = "barcode_arrs_cust2"
id_pattern = "CUST%02i"
compatible_kits = ["MY-CUSTOM-BARCODES"]
first_index = 1
last_index = 2
kit = "CUST"
normalised_id_pattern = "barcode%02i"
scoring_function = "MAX"
barcode1_pattern = "CUST%02i"
barcode2_pattern = "CUST%02i"
Note that we set bothbarcode1_patternandbarcode2_patternto the same value. This means:- We are going to search for barcodes at the rear of the strand (because barcode2 is set).
- The rear barcodes are based on the same barcodes used in the front.
Note: If the range of barcode indices includes values greater than 99, ensure that sufficient digits are specified in each of the pattern fields. For example, if you have barcodes from 1 to 384, the arrangements section would contain pattern fields containing
%03i, like this:[arrangement] name = "barcode_arrs_cust2" id_pattern = "CUST%03i" compatible_kits = ["MY-CUSTOM-BARCODES"] first_index = 1 last_index = 384 kit = "CUST" normalised_id_pattern = "barcode%03i" scoring_function = "MAX" barcode1_pattern = "CUST%03i" barcode2_pattern = "CUST%03i"
Step 4: Add the new barcodes to the FASTA file
Open thecustom_barcodes.fastafile created during step 2 and add your barcode sequences in with names matching theCUST%02ipattern used in the arrangement file:```
CUST01
AAAAAAAGCTCGCTCGCTCGAGATTTTTTT
CUST02
AAAAAAACGGTAAATTGGCATTATTTTTTT
``
<br>
**Step 5: Run
guppy_barcoder \
--input_path [path_to_input_fastq_files] \
--save_path [path_to_output_directory] \
--data_path ~/mydata/barcoding \
--barcode_kits MY-CUSTOM-BARCODES
- In
-
Context-specific barcode specification
When specifying a LAMP arrangement, context-specific barcodes are supported. For example, consider a LAMP configuration file as follows:
[loading_options]
barcodes_filename = "barcodes_masked.fasta"
lamp_targets_filename = "lamp_targets.fasta"
double_variants_frontrear = true
[arrangement]
name = "barcode_arrs_lamp_example"
id_pattern = "LAMP%02i"
compatible_kits = ["MY_CUSTOM_LAMP_KIT"]
first_index = 1
last_index = 8
kit = "LAMP"
normalised_id_pattern = "FIP%02i"
barcode1_pattern = "LM%02i"
lamp_masks = ["CONTEXT1","CONTEXT2","CONTEXT3"]
Normally, this kit would be expanded to use barcodes
LM01toLM08from thebarcodes_masked.fasta, no matter which context is being used. However, there are situations where having a context-specific barcode may be desirable. If a specific barcode is required to replaceLM01forCONTEXT2, it can be added to thebarcodes_masked.fastaas follows:```
LM01
ACGTATCTCA
```

