-
Recommended analysis pipeline
The recommended workflows for the bioinformatics analyses are provided by the ARTIC network and are documented on their web pages at https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html.
The reference guided genome assembly and variant calling are also performed according to the bioinformatics protocol provided by the ARTIC network. Their best practices guide uses the software contained within the FieldBioinformatics project on GitHub.
This workflow uses only the basecalled FASTQ files to perform a high-quality reference-guided assembly of the SARS-CoV-2 genome. Sequenced reads are re-demultiplexed with the requirement that reads must contain a barcode at both ends of the sequence (this only applies to the Classic and Eco PCR tiling of SARS-CoV-2 protocols but not the Rapid Barcoding PCR tiling of SARS-CoV-2), and must not contain internal barcodes. The reads are mapped to the reference genome, primer sequences are excluded and the consensus sequence is polished. The Medaka software is used to call single-nucleotide variants while the ARTIC software reports the high-quality consensus sequence from the workflow.
-
Expected results
Here, results are shown based on human coronavirus 229E spiked into 100 ng of human RNA derived from GM12878 cell line. 10 pg–0.001 pg of viral RNA obtained from ATCC was spiked into the human RNA and human-only and reverse transcription negative controls were carried through the prep to sequencing. Every sample underwent 30 and 35 cycles of PCR to determine sensitivity and specificity guidelines, as well as the expected amplicon drop-out rate for each sample.
Note: The viral RNA from ATCC is generated from cell lines infected with human coronavirus 229E. The RNA supplied is total RNA extracted from the cell lines and includes both human and viral RNA. Therefore, the levels of sensitivity are likely to be higher than those reported here.
-
Sample balancing
The graph below shows the expected sequence balancing if the protocol is followed. Here, equal masses went into the end-prep and native barcode ligation prior to pooling by equal mass for adapter ligation.
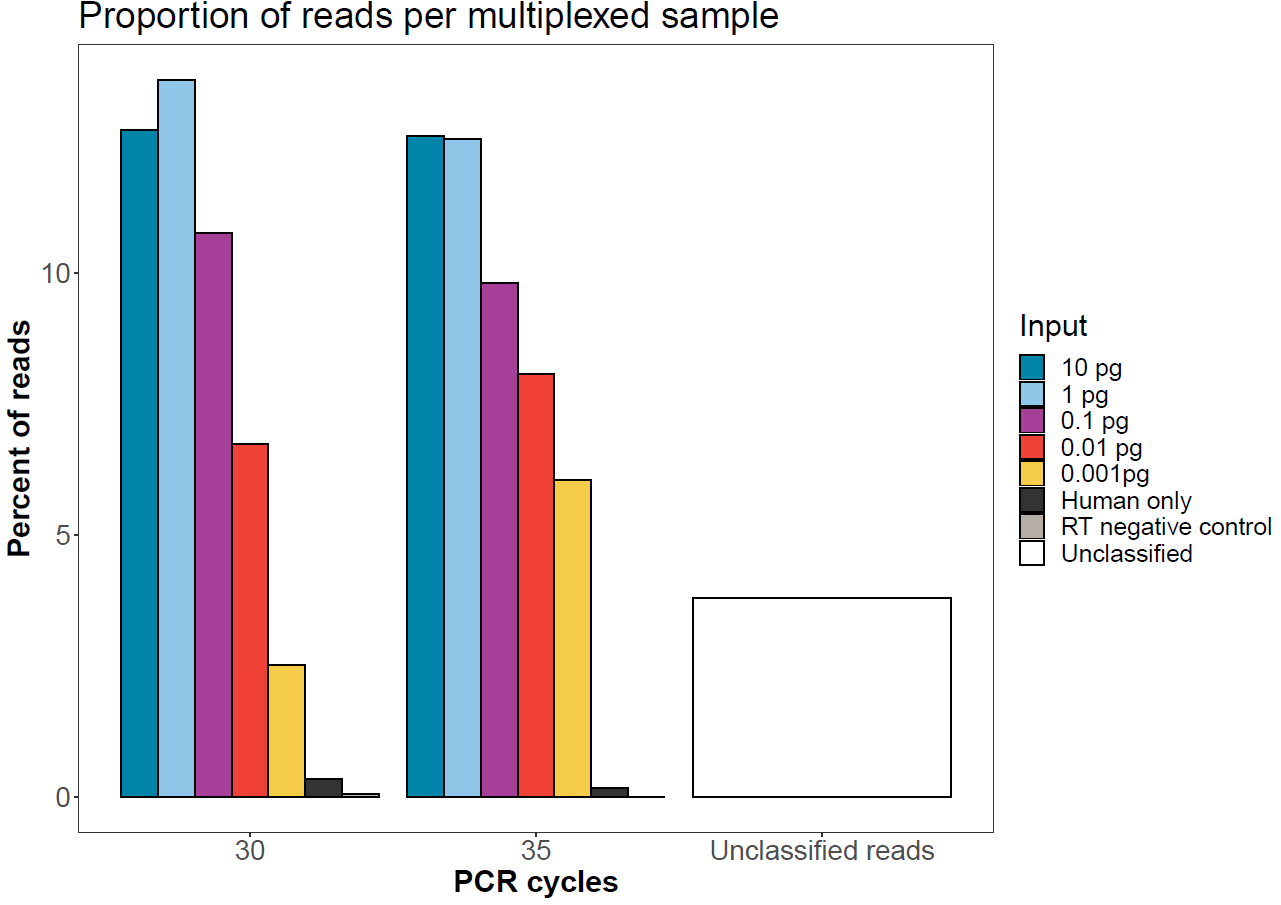
Figure 3. Number of reads per sample after native barcode demultiplexing in MinKNOW. All 14 samples were run on a single flow cell. -
On-target rate
Sequences from each demultiplexed sample were aligned to the human coronavirus 229E genome using minimap2. The proportion of primary alignments per sample are reported below.
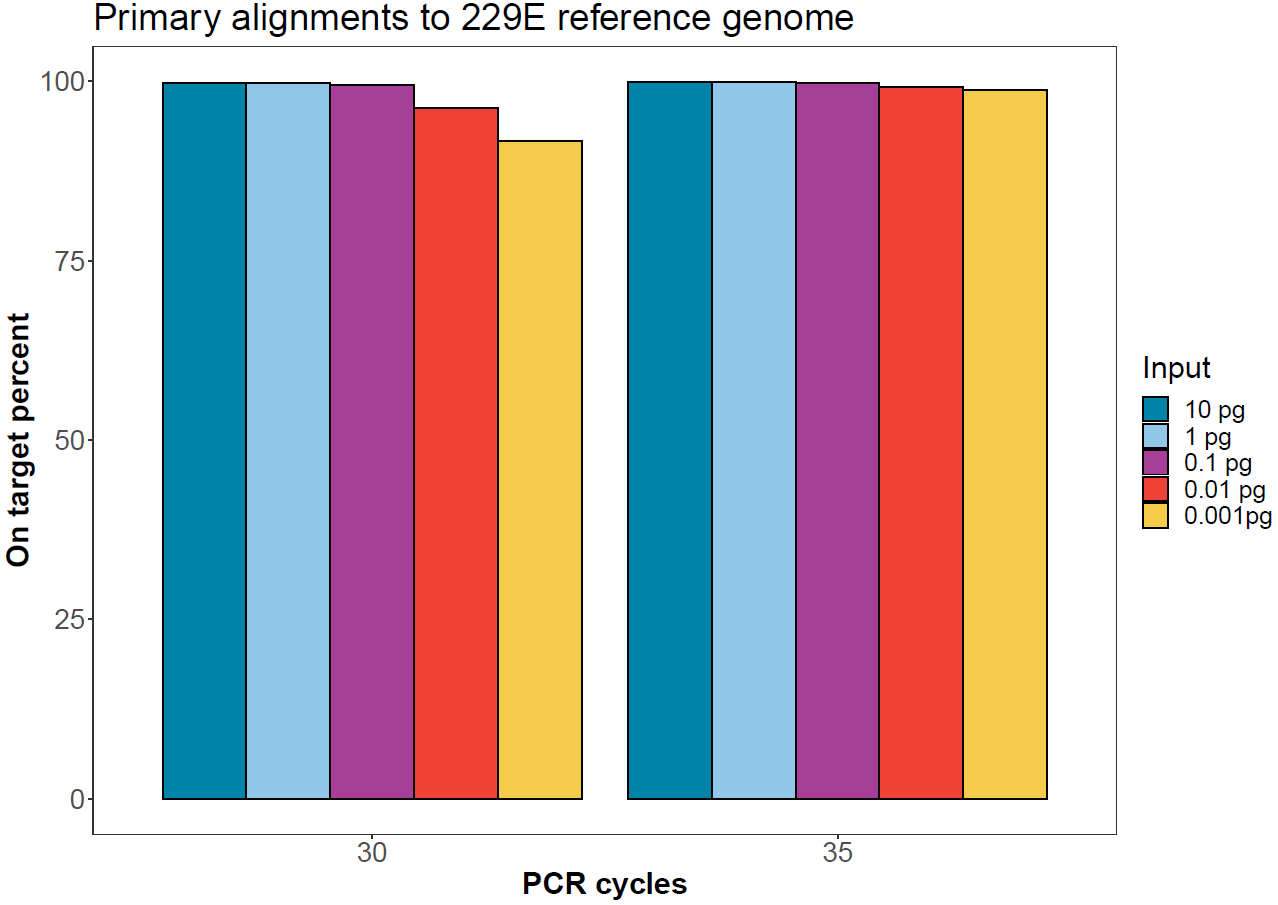
Figure 4. Proportion of reads for each sample aligning to the human coronavirus 229E reference genome. -
Assessment of negative controls
After 12 hours of sequencing, the number of reads from the negative control samples aligning to the viral reference genome is shown in the graph below and is compared with the absolute number of sequences aligning to the lowest input (0.001 pg).
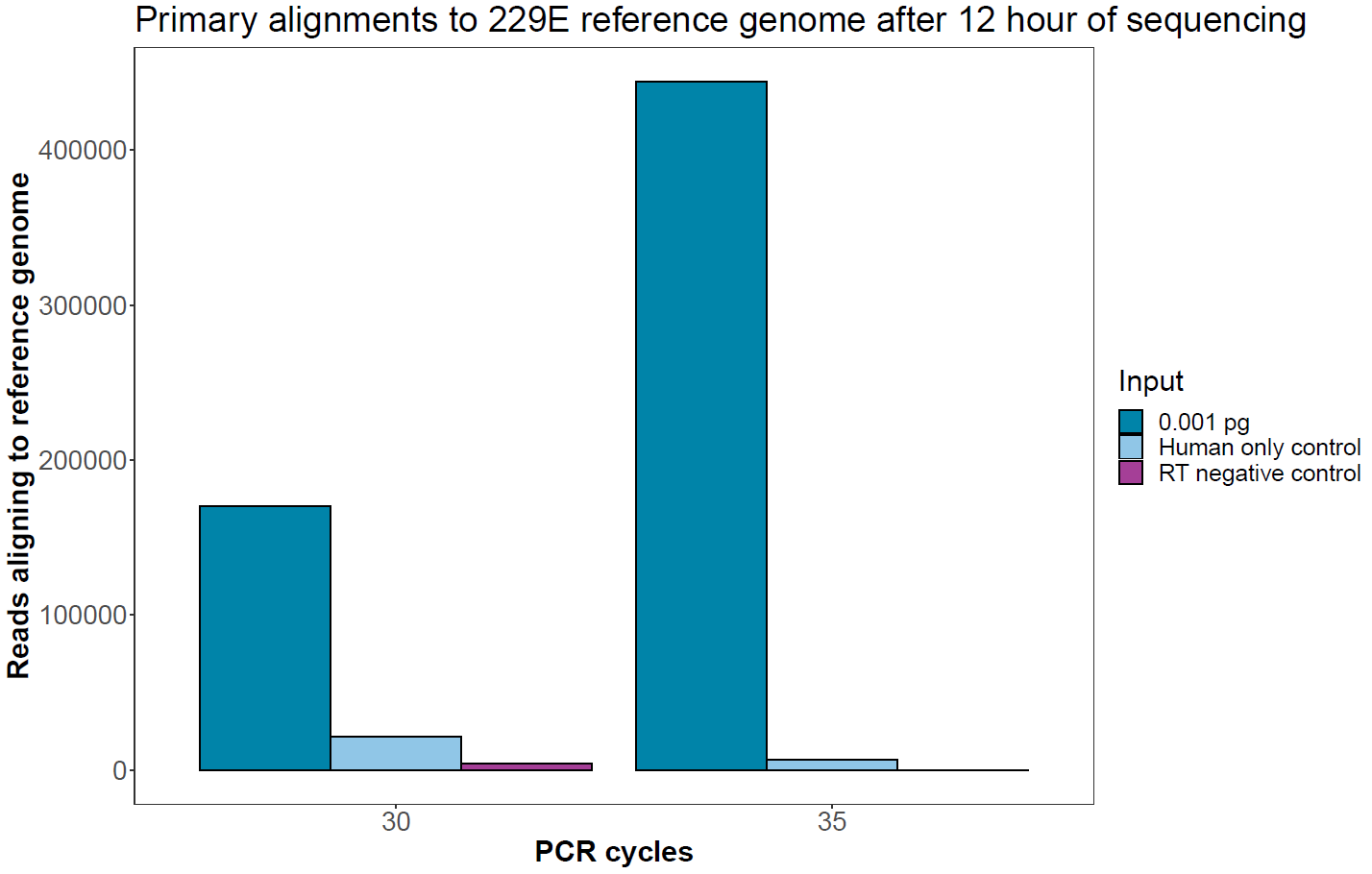
Figure 5. Absolute number of reads aligning to the human coronavirus 229E reference genome in the negative controls compared with the lowest input of viral RNA. Sequencing was carried out for 12 hours to pick up low levels of sequences assigned to barcodes representing these samples. -
Target coverage for different PCR cycles and viral load
To assess the impact of PCR dropout with lowering input viral load and increasing PCR cycles, Mosdepth was used to calculate the proportion of the viral genome covered to different depth levels. These numbers were calculated after 12 hours of sequencing with 14 samples multiplexed.
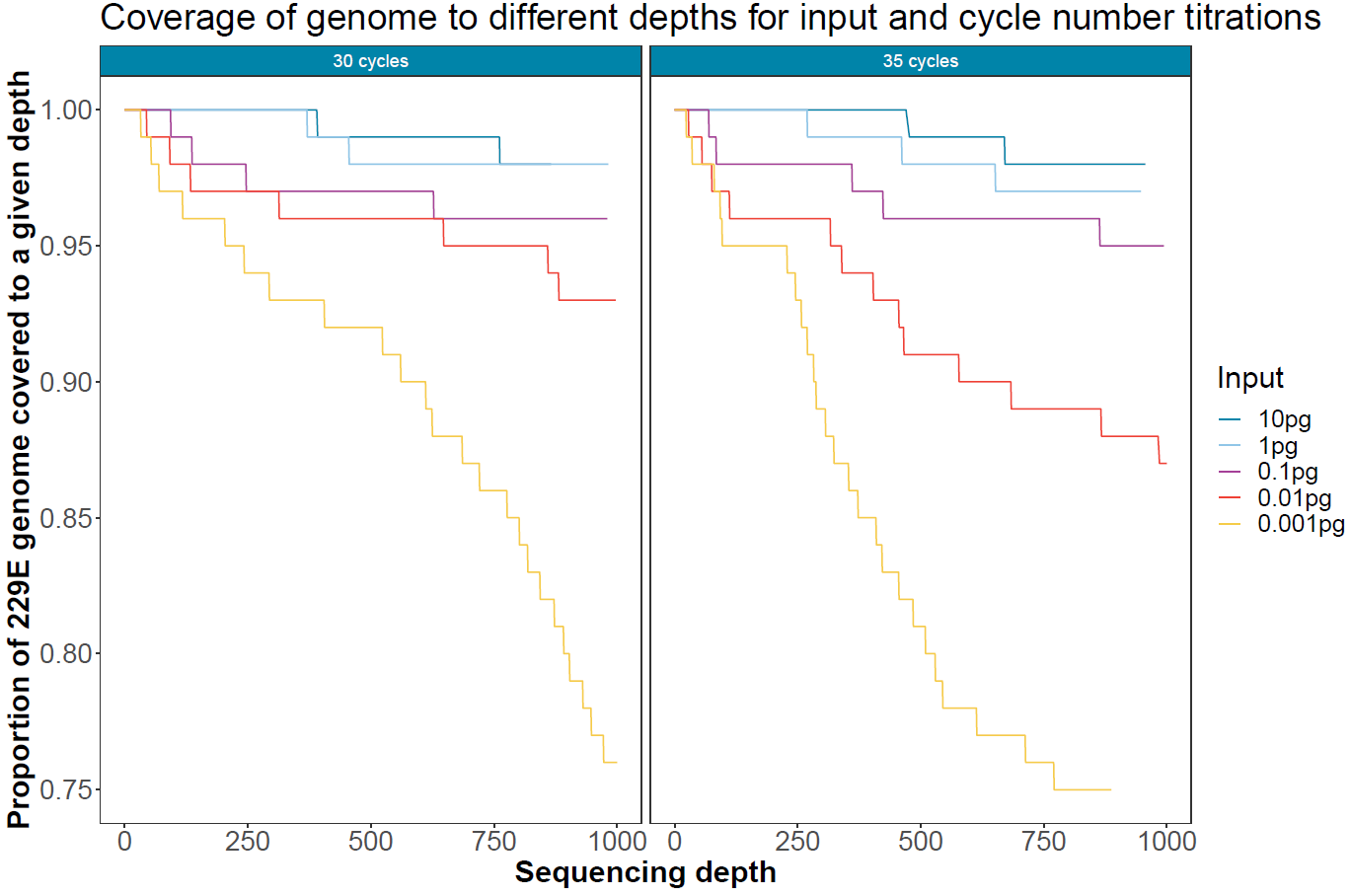
Figure 6. Coverage and depth of the human coronavirus 229E genome for different input quantities of viral RNA and different cycle numbers after 12 hours of sequencing on a single flow cell. -
How much sequencing is required?
This is unknown in real clinical samples. The graph below can be used to determine the proportion of the genome that could be covered to a given depth with different numbers of reads (30 cycles) at different input amounts in a background of 100 ng human RNA.
Note: this is absolute depth.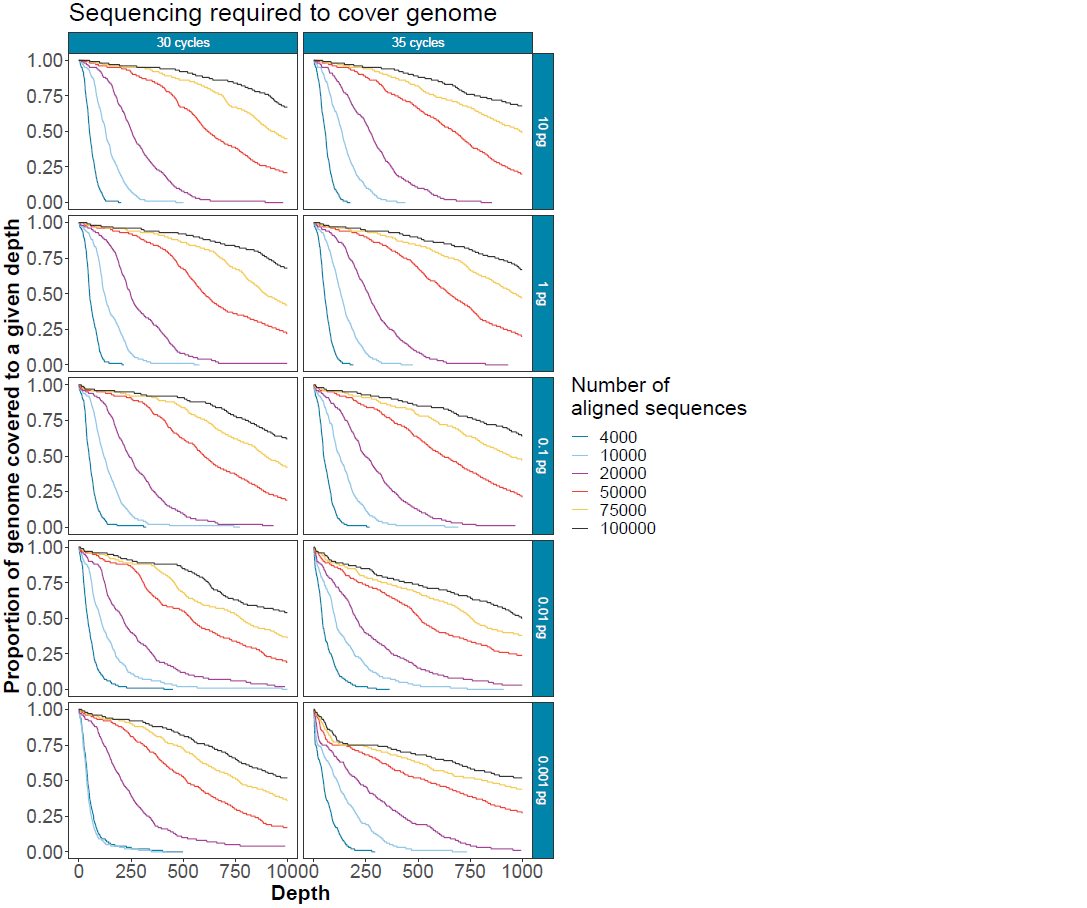
Figure 7. Subsampled sequences to give an indication of the depth of sequencing achievable covering different amounts of the human coronavirus 229E genome. Input quantities and cycle number titrations show that high cycle numbers should be avoided where possible to minimise amplicon drop out.This protocol provides amplification of low copy number viral genomes in a tiled method with low off-target amplification and minimal cross-contamination between samples. With <60 copies per reaction (0.001 pg viral input) in 100 ng background human RNA, under ideal circumstances, one should expect to cover >75% of the targeted genome at a depth of 200X within under 50,000 reads in the samples with the lowest viral titre and <20,000 reads in those with a higher viral titre.




