-
Introduction to DNA quality control
It is important that you check your input DNA for quality before beginning library preparation. Low molecular weight, incorrectly quantified and/or contaminated DNA (e.g. salt, EDTA, protein, organic solvents) can have a significant impact on downstream processes and ultimately, your sequencing runs. Below are some guidelines for how to check the DNA quality to ensure the highest possible throughput.
Access to laboratory equipment is not always possible in field conditions, so we recommend that you optimise extraction and purification in the laboratory before doing fieldwork.
To obtain similar results to those generated in the Lambda Control protocol, use high quality, pure genomic DNA (gDNA). We have had good results with DNA which meets the following criteria:
- Purity as measured using Nanodrop – OD 260/280 of 1.8 and OD 260/230 of 2.0–2.2
- Average fragment size, as measured by pulsed-field gel analysis ( >30 kb)
- Input mass, as measured by Qubit – 1 µg
For long-term storage of high molecular weight (HMW) gDNA we recommend the use of TE buffer.
There are established methods for generating high quality HMW gDNA which should be used when preparing gDNA for sequencing on the MinION Mk 1B. Several of these methods are described in detail in Sambrook J, and Russell DW, (2001) Molecular Cloning: a Laboratory Manual. 3 rd Edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
-
In order to maximise sequencing yield, it is important that the nanopores are kept filled with DNA to minimize the time they are idle between strands. The less material goes into the flow cell, the fewer “threadable ends” will be present to be captured by the pores. Therefore, the pores will be searching for molecules for longer, and if the pores are not always sequencing, throughput could be compromised. We have found that in order to keep the pores full, the current R9.4.1 pore requires about 5-50 fmol of good quality library put into the flow cell, but no more than 600 ng. If you are using the Flongle for sample prep development, we recommend loading 3-20 fmol instead. The relationship between input and throughput on R9.4.1 with the Ligation Sequencing Kit (SQK-LSK109) is shown below.
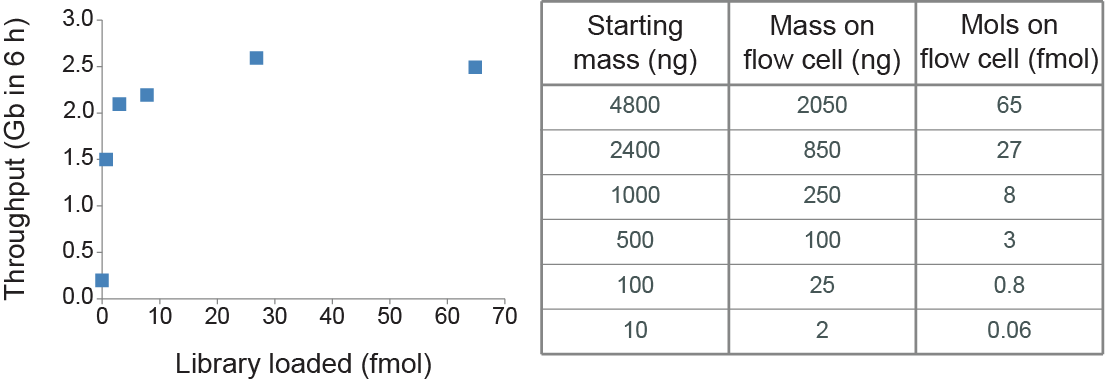
All throughputs shown are from MinION Mk1B. The data was generated using the Lambda control DNA, and the molarity conversion assumes a fragment size of ~48.5 kbInput DNA quantity and fragment length
To ensure you are starting with the correct amount of material, the sample should be properly quantified in terms of mass and fragment length. We recommend the Qubit fluorometer to determine the mass of the sample, and to determine the fragment size of the sample you could use: gels (e.g. Bio-Rad CHEF system), Agilent devices (e.g. Bioanalyzer or Tapestation), or FEMTO Pulse from AATI.We have also observed that sequencing throughput depends partly on fragment length: shorter fragments take less time to be processed/sequenced and therefore the pores return to the open state more often than when sequencing longer fragments.
Our input recommendations are:
- Samples with a wide distribution of fragment sizes, e.g. gDNA: start with 1 μg of material for the Ligation Sequencing Kit, or 400 ng of material for the Rapid Sequencing Kit
- Short fragment libraries, e.g. amplicons or cDNA: start with 100-200 fmol
- Some kits which have a PCR step included as part of the protocol can be used with lower DNA inputs. Please refer to individual library prep protocols for detailsIf you are unable to quantify your input DNA mass, please use the table below as a guide. Then, take forward the appropriate amount of DNA based on the average fragment length and known concentration.
Mass No. of moles if fragment length = 2 kb No. of moles if fragment length = 8 kb No. of moles if fragment length = 50 kb 10 μg 7.7 pmol 1.9 pmol 308 fmol 5 μg 3.9 pmol 963 fmol 154 fmol 3.5 μg 2.7 pmol 674 fmol 108 fmol 2 μg 1.5 pmol 385 fmol 62 fmol 1.5 μg 1.2 pmol 289 fmol 46 fmol 1 μg 770 fmol 193 fmol 31 fmol 500 ng 385 fmol 96 fmol 15 fmol 400 ng 308 fmol 77 fmol 12 fmol 200 ng 154 fmol 39 fmol 6.2 fmol 100 ng 77 fmol 19 fmol 3.1 fmol 30 ng 23 fmol 5.8 fmol 0.9 fmol 10 ng 7.7 fmol 1.9 fmol 0.3 fmol 10 pg 0.0077 fmol 0.009 fmol 0.0003 fmol Fragmentation at low inputs
We grade our library preparation kits and sample extraction protocols using a 3 star system:MinION/GridION throughput per flow cell PromethION throughput per flow cell 1 star 0-1 Gbases in 6 h (1-4 Gbases in 48 h) 0-5 Gbases in 6 h (5-20 Gbases in 48 h) 2 star 1-2 Gbases in 6 h (4-8 Gbases in 48 h) 5-10 Gbases in 6 h (20-60 Gbases in 48 h) 3 star 2-3+ Gbases in 6 h (8+ Gbases in 48 h) 10-15+ Gbases in 6 h (60+ Gbases in 48 h) We find that 1000 ng (or even 500 ng) of unfragmented gDNA generates 3 star performance. However, at ~100 ng, the performance begins to suffer, dropping from 3 to 2 star. However, it is possible to regain 3 star performance by g-TUBE fragmentation of the sample. When the input drops to below 100 ng (e.g. ~10 ng), then g-TUBE fragmentation may not be sufficient to regain 3 star, or even 2 star performance. Here, we would recommend using our range of PCR sequencing kits to generate more template and increase throughput.
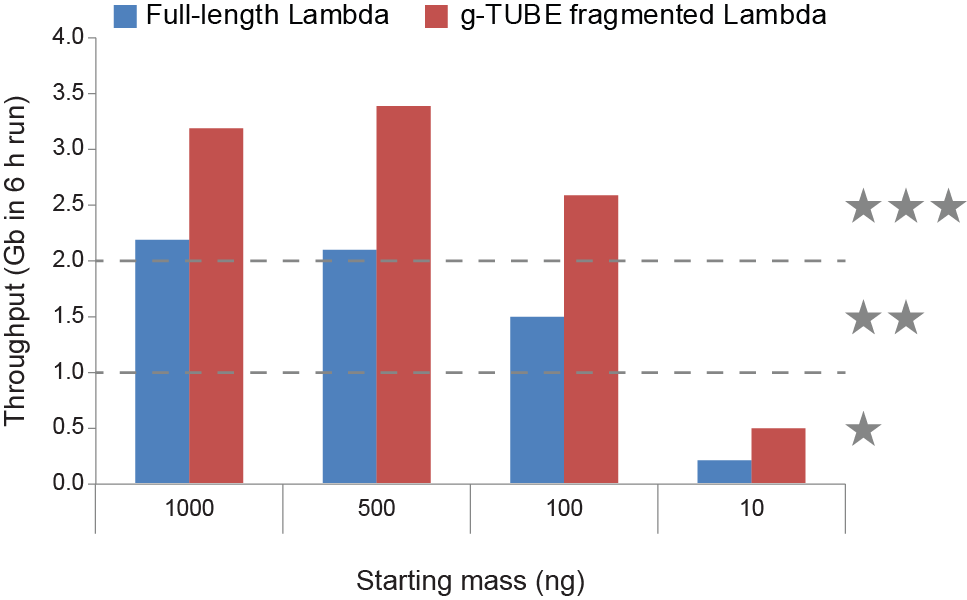
All throughputs shown are from MinION -
Correct quantification
The majority of RNA should be removed by RNase digestion. We have found RiboShredder (Epicentre RS 12500) to be particularly effective. However, since Riboshredder is being discontinued, you can use the RNase Cocktail Enzyme Mix (ThermoFisher, AM2286) instead.
Please be aware that certain RNase treatments can lead to digestion of DNA, as well as RNA.We recommend that the DNA stock is quantified using Qubit analysis. A Qubit measures DNA specifically. Even after RNase digestion, residual RNA is a common contaminant in gDNA preparations and is not well identified by Nanodrop measurements. Incorrect quantification could mean that you will proceed with less DNA than intended, resulting in poor performance. Also, contamination from bases (dNTPs and NTPs) will interfere with Nanodrop measurements. Therefore, we recommend that Qubit is used for all quantification measurements (i.e. after all clean-up steps).
Additionally, high concentration, high molecular weight DNA preparations (and those with heavy RNA contamination) can lack homogeneity, which will give rise to inaccurate quantification. If you encounter this with your RNase-treated DNA sample, we recommend that you dilute the DNA further with TE, and that you rotate the tube gently until the suspension is homogeneous. Vortexing the DNA or pipetting up and down will cause shearing, which will limit the fragment sizes available to the nanopore.
-
Assessing DNA quality
- Chemical impurities such as detergents, denaturants, chelating agents and high concentrations of salts should be avoided as these may affect the efficiency of enzymatic steps. Please refer to the Contaminants document for more details.
- Other contaminants such as single stranded DNA, RNA, proteins and dyes may also reduce the efficiency of steps in the library preparation.
- The quality of DNA may be assessed by Nanodrop (for samples with concentration >20 ng/µl).
- We recommend that sample DNA has a 260/280 ~ 1.80 and a 260/230 ~2.0-2.2.
- A 260/280 which is higher than ~1.8 indicates the presence of RNA.
- A 260/280 which is lower than ~1.8 can indicate the presence of protein or phenol.
- A 260/230 significantly lower than 2.0-2.2 indicates the presence of contaminants, and the DNA may need additional purification.
In the Nanodrop trace shown below, Sample 1 had a 260/230 of ~1.0 and the resulting library performed badly in a sequencing run. If additional purification is not possible, amplification of the library by PCR can be performed to improve library cleanliness.

-
Assessing molecular weight
Nanopore sequencing devices generate reads that reflect the lengths of the fragments loaded into the flow cell. To have control over the size of the fragments generated in the library prep, it is important to begin with high molecular weight (HMW) DNA.
The shearing of HMW DNA can be minimised by:
- Using wide-bore pipette tips to handle the gDNA
- Mixing gently but thoroughly by flicking the tube, as opposed to vortexing or pipetting
- Avoiding unnecessary freeze-thaw cycles
- Avoiding pH <6 and >9
- Avoiding high temperatures, which can lead to degradation
Conventional agarose gels cannot resolve DNA fragments greater than 15–20 kb, but the molecular weight of starting material can be measured by pulsed-field gel analysis.
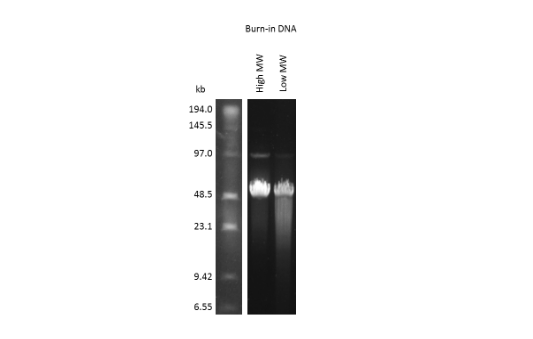
The figure shows two samples of Lambda phage DNA: one of intact high molecular weight fragments and one containing a significant proportion of low molecular weight fragments.
Low % agarose gel analysis can be used to detect substantial degradation/shearing:
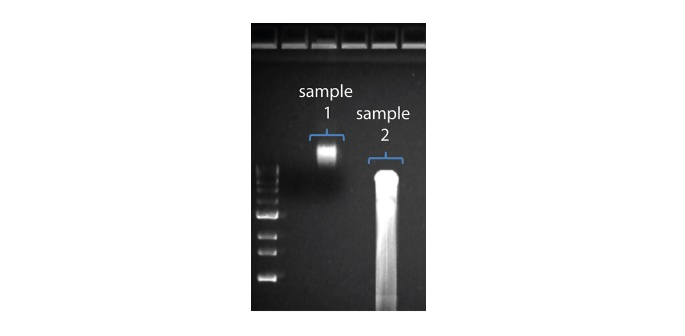
The figure shows two samples of input DNA: Sample #1 is of high molecular weight and sample #2 is of lower molecular weight and has sheared.
-
Assessing fragmentation
Post-fragmentation, the quality of the fragmented material may be assessed by different methods e.g. Agilent Bioanalzyer.

The figure above shows successful (sample 1) and unsuccessful (sample 2) fragmentation, demonstrated by an Agilent Bioanalyzer trace of two DNA samples. Sample #2 contains a substantial proportion of low molecular weight fragments. This is possibly as a result of improper fragmentation, or these low MW fragments may have been present in the input sample.
-
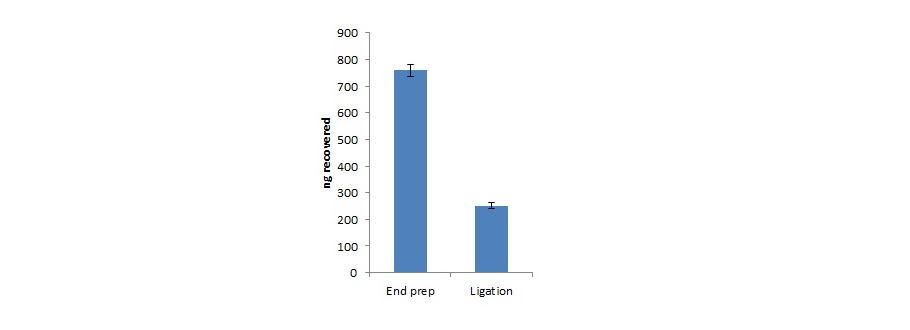
Assessing DNA recovery during library preparation
We recommend performing QC checks at certains points in the library preparation process. This allow users to know how much DNA they have at each stage. Starting with 1 µg (as measured by Qubit), the graph below outlines what should be expected after end repair, dA-tailing and adapter ligation with the Ligation Sequencing Kit. Throughput of a library that follows this quantification is expected to give good throughput.