-
How to start sequencing
The sequencing device control, data acquisition and real-time basecalling are carried out by the MinKNOW software.
We recommend basecalling with the high accuracy (HAC) basecaller in real-time with BAM selected as output type using the P2 Solo or P24/P48 device.
A BAM file must be generated as this is required for input into the wf-human-variation workflow. We recommend aligning the BAM output after live basecalling to prevent slowing sytem processing or to provide a reference genome along with the unaligned BAM file in the wf-human-variation workflow. Using mapped BAM as input, the workflow will take 1-2 hours. If unmapped BAM is used as input, the workflow will take approximately 5-8 hours. Further information is available in Downstream analysis.
Refer to the links below containing the detailed instructions for setting up the device and sequencing run:
PromethION 24 and 48: "Starting a sequencing run with PromethION 24 and 48"
PromethION 2 Solo: "Starting a sequencing run on PromethION 2 Solo"
Below are the recommended sequencing parameters for MinKNOW.
-
MinKNOW settings for 30kb human sample on PromethION
For inputs of 30 kb, we recommend increasing the run time to 100 hours to accommodate for the two flow cell washes, using the modified bases option for basecalling and ensuring a BAM output is selected. All other sequencing parameters can be kept to their default settings.
Below are our current recommendations:Positions
Flow cell position: [user defined]
Experiment name: [user defined]
Flow cell type: FLO-PRO114M
Sample ID: [user defined]
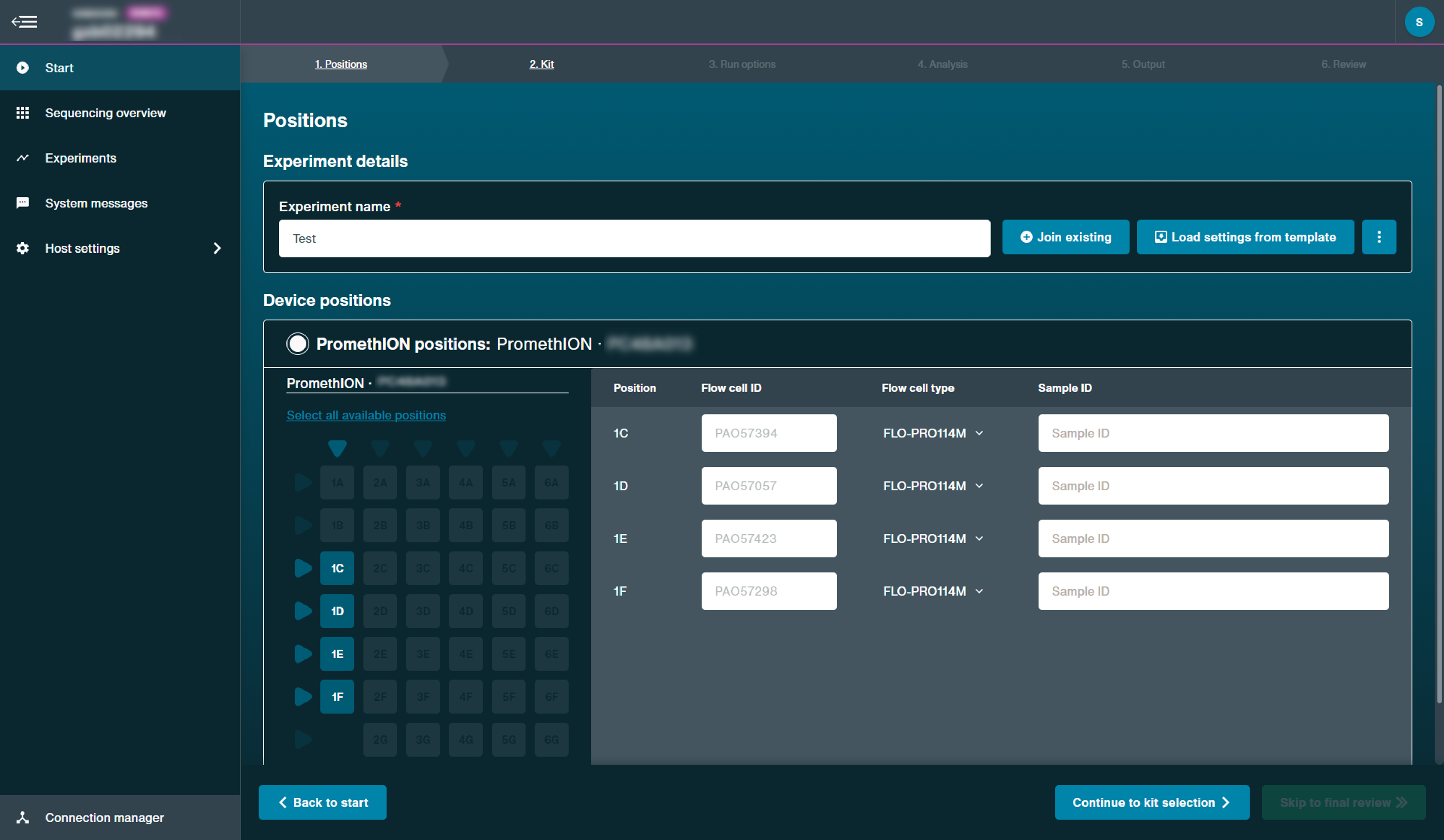
Kit
Kit selection: Ligation Sequencing Kit (SQK-LSK114)

Run options
Run limit: 100 hours
Minimum read length: 200 bp [default]
Adaptive sampling
Enrich or deplete sequening: Off [default]
Barcoding balance: Off [default]Advanced options
Active channel selection: On [default]
Time between pore scans: 1.5 [default]
Reserve pores: On [default]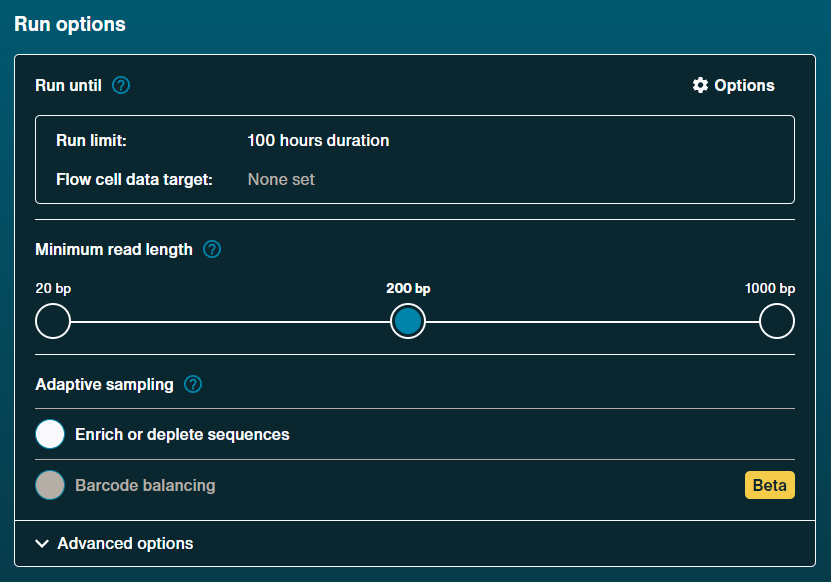
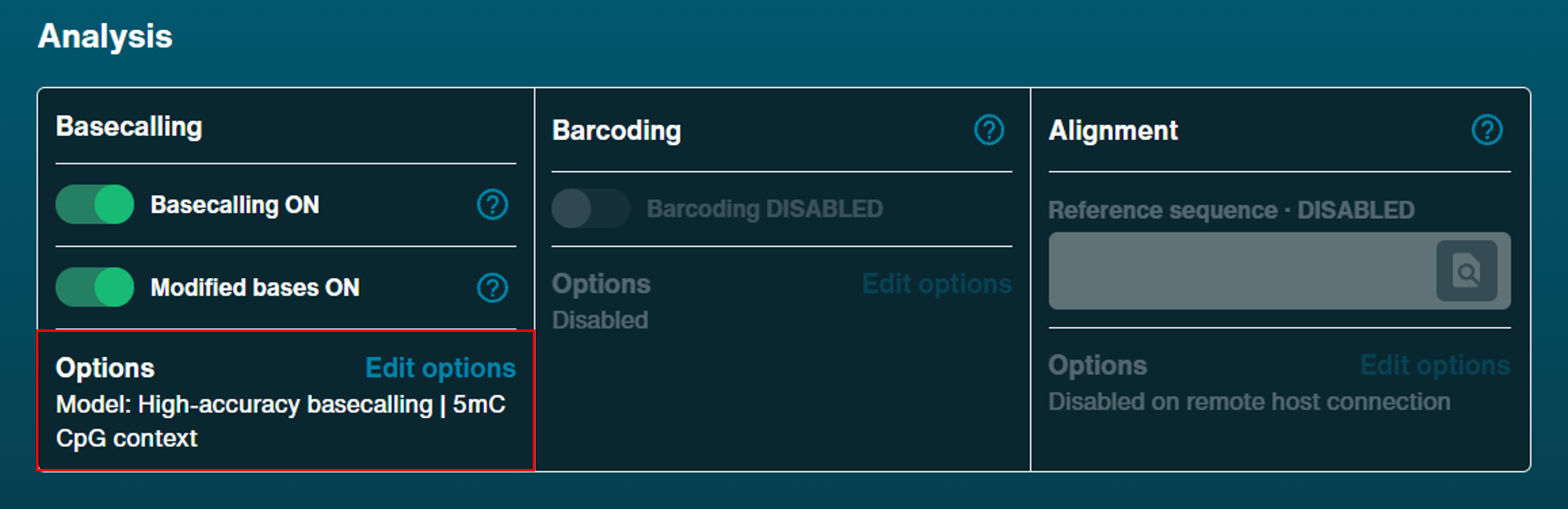
Analysis
Basecalling
Basecalling: On [default]
Modified bases: On with '5mC' selected
Model: High-accuracy basecalling (HAC) [default]Barcoding
Barcoding: Disabled [default]Alignment
Reference sequence: Off [default]Note: We do not currently recommend live alignment during sequencing, as it can slow down system processing.
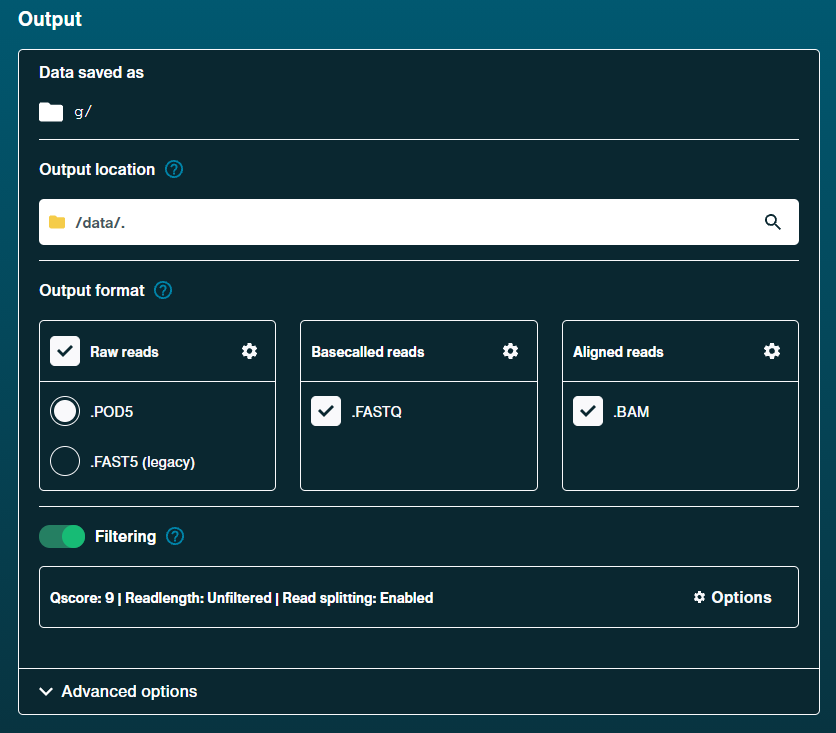
Output
Output format
.POD5: On [default]
.FASTQ: On [default]
.BAM: OnFiltering: On [default]
Qscore: 9 [default]
Readlenth: Unfiltered [default]
Read splitting: Enabled [default]