-
Recommended analysis pipeline
The analysis of the FASTQ format sequence data is performed using a Nextflow workflow called Influenza Typing Workflow (wf-flu). The use of the Nextflow software has been integrated into the EPI2ME Labs software that we recommend for running our downstream analysis methods.
Alternative methods for downstream analysis are available using your device terminal or command line, however we only suggest this for experienced users.
The workflow processes the basecalled and demultiplexed DNA sequence data output generated by MinKNOW:
- The sequences are filtered for a minimum length and quality thresholds (200 nucleotides and Q9 respectively) prior to sequence alignment to the CDC multi-fasta Influenza reference.
- The alignment is performed using the Minimap2 software.
- Depth of coverage across the mapped sequences is measured using Samtools before genetic variants are called using Medaka.
- A coverage-masked consensus sequence is prepared for each sample using bcftools.
- The influenza strain typing is then performed using the abricate software with an insaflu database.
The influenza strains included in the database are listed in the project documentation pages for the Influenza Typing Workflow.
The workflow returns a per-run HTML-format summary report along with a CSV file of typing results. Additional files that include mapping BAM files and VCF files of Medaka variants are also included in the workflow output.
For more information, please refer to the Influenza workflow blog.
-
Software set-up and installation
The EPI2ME Labs application provides a clean interface to accessing bioinformatics workflows, and is our recommended method in performing your post-sequencing analysis.
Follow the instructions in the EPI2ME Labs Installation guide to install the application on your device.
For more information on how to use EPI2ME Labs, refer to the EPI2ME Labs Quick Start guide.
-
Installing and updating the wf-flu workflow in EPI2ME Labs:
Ensure you have installed the wf-flu workflow prior to the first analysis set-up.
In the EPI2ME Labs home page, scroll down to the "Install workflows" section and click on epi2me-labs/wf-flu:
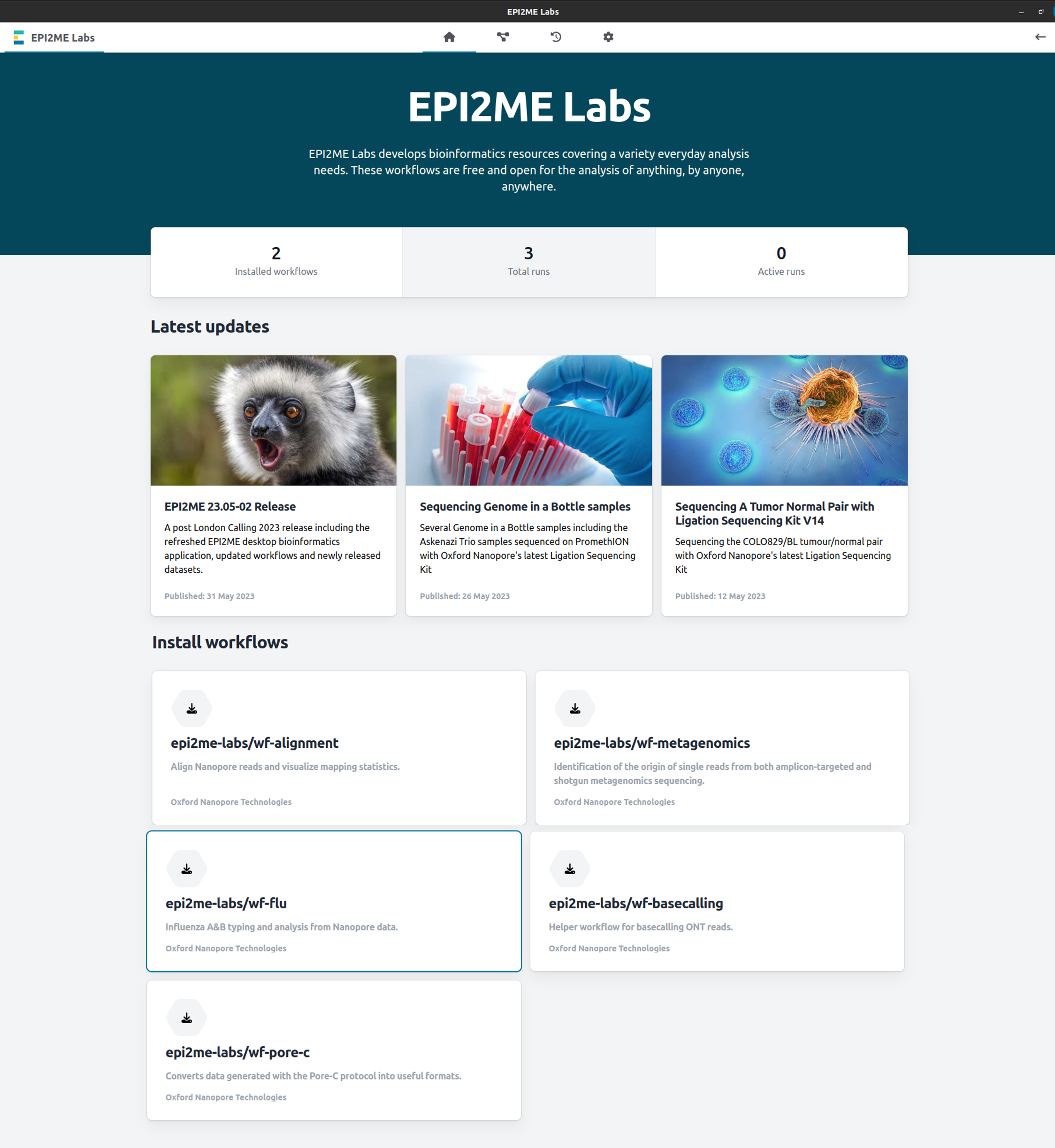
If you have already installed the wf-flu workflow, ensure you are using the latest version.
Updating the workflow can be done directly through EPI2ME Labs by navigating to the wf-flu workflow page and clicking Update Workflow:
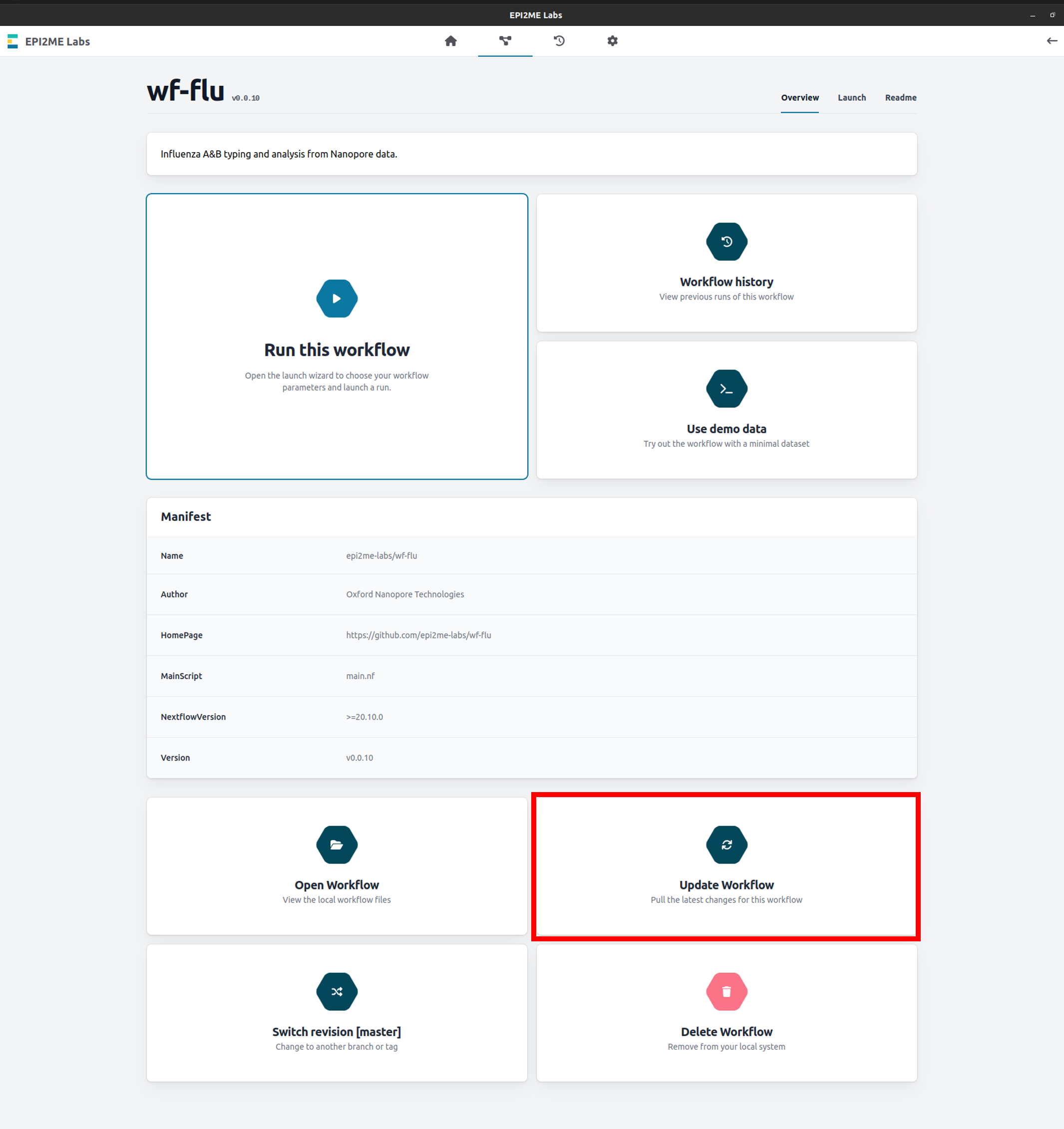
-
Demultiplexing of multiple barcoded samples
The wf-flu analysis requires FASTQ sequence data that has already been demultiplexed.
Reads will be demultiplexed during sequencing if you are following the recommended "Required settings in MinKNOW". However, demultiplexing can also be done post-sequencing using the MinKNOW software.
For more information and guides on demultiplexing using MinKNOW, refer to the "Post-run analysis" section in our MinKNOW Protocol.
The expected input for wf-flu is a folder of folders as shown below. Each of the barcode folders should contain the FASTQ sequence data and files may either be uncompressed or gzipped.
$ tree -d FluFastq/FluFastq/├── barcode01├── barcode02├── barcode03├── barcode04├── barcode05├── barcode06└── unclassified -
Running a Flu analysis using EPI2ME Labs
-
Open the EPI2ME Labs application on your device.

-
Open the "Workflows" tab in the EPI2ME Labs application and click on the "wf-flu" workflow:
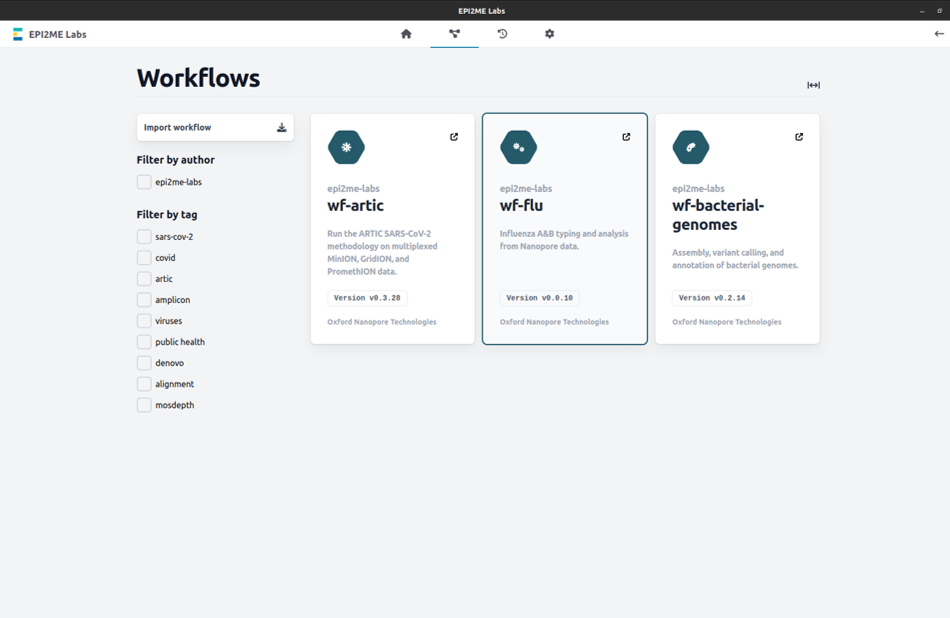
-
In the "wf-flu" workflow page, select "Run this workflow" to open analysis set-up:
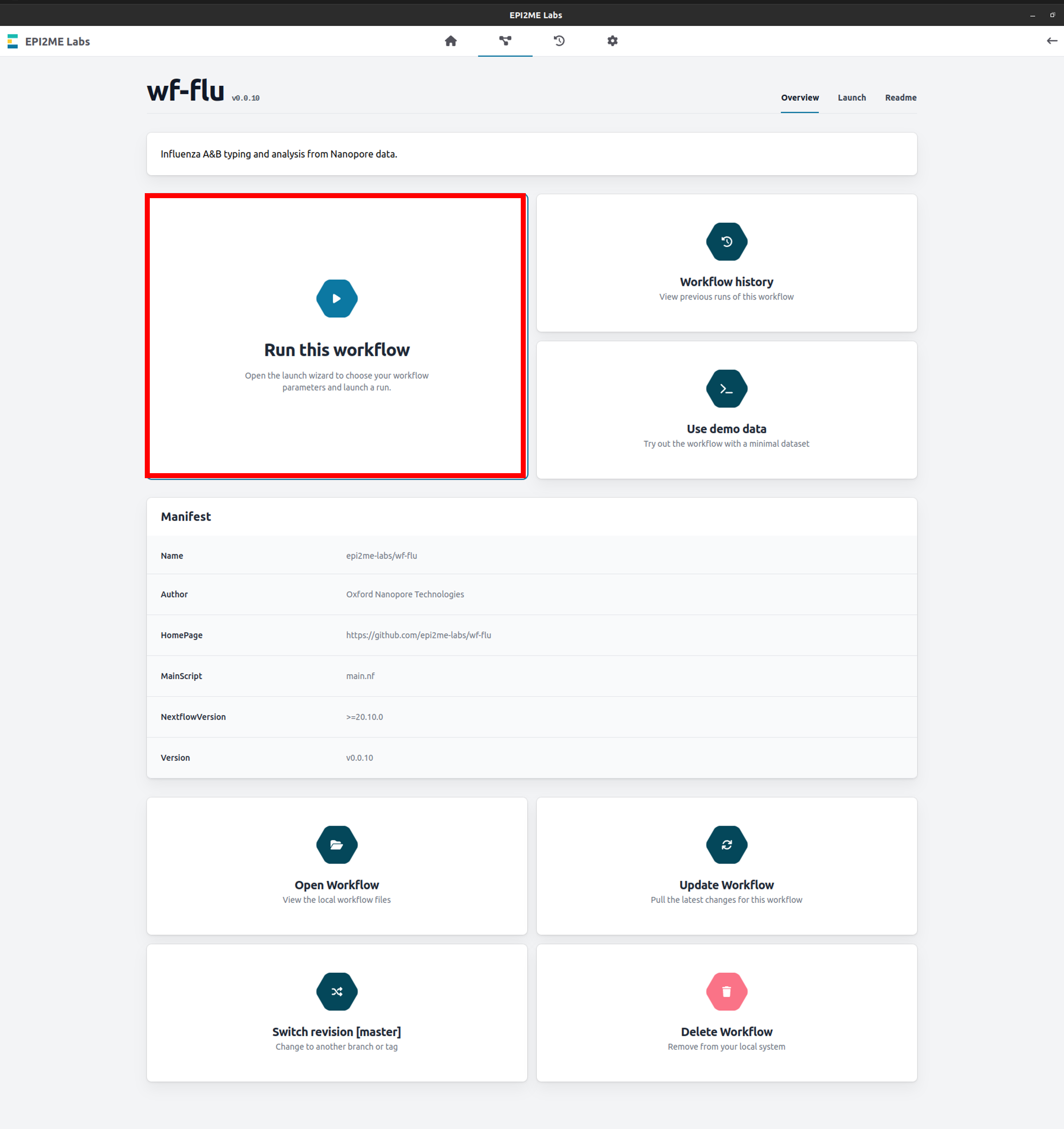
-
Complete the wf-flu run set-up:
Select your data input file location. Please note, this folder must contain the demultiplexed FASTQ files of your sequencing run.
Set the basecaller cgf to the basecalling model used in your sequencing run.
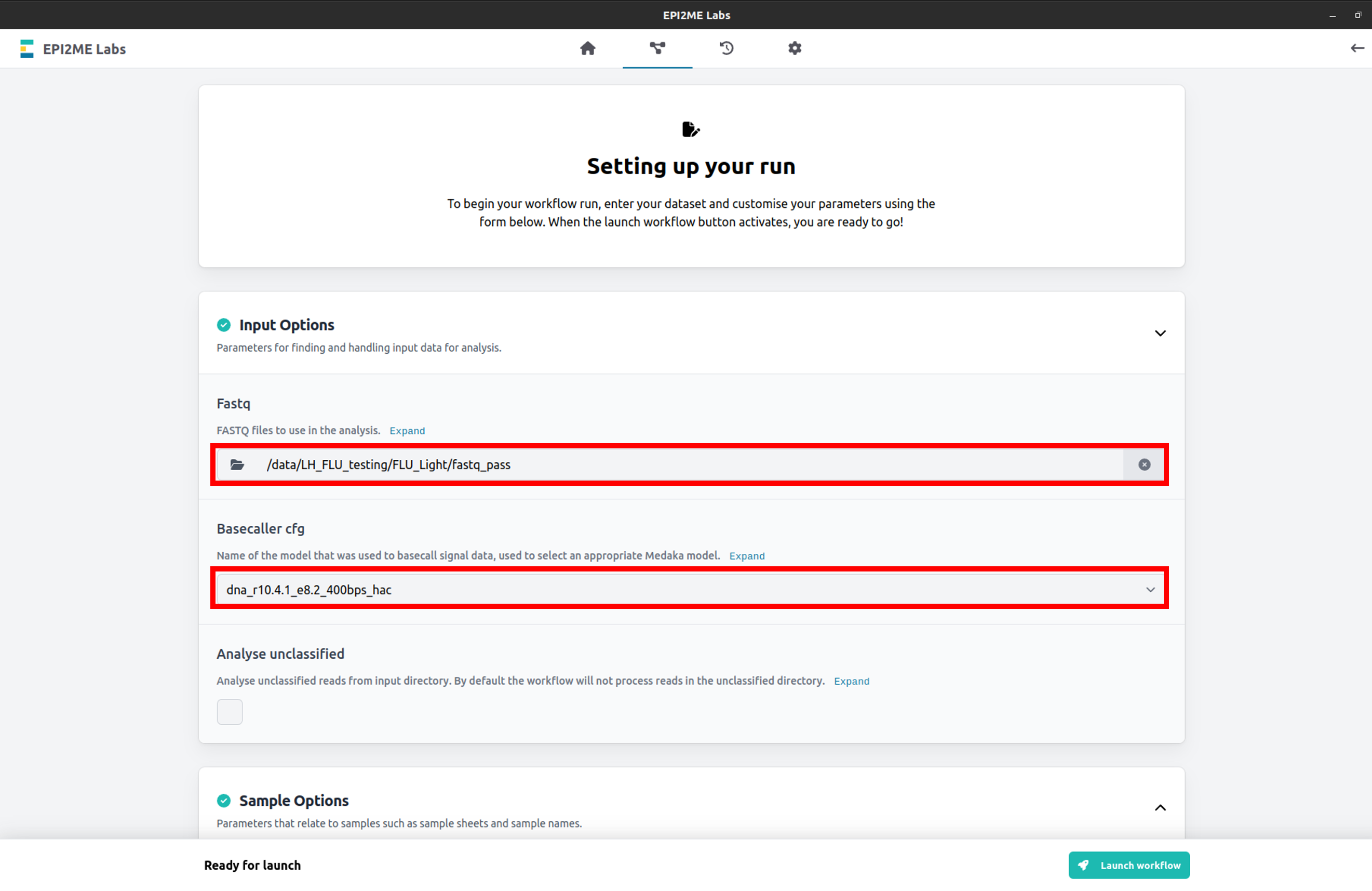
Expand the Extra configuration tab and set the Run name for your wf-flu analysis.
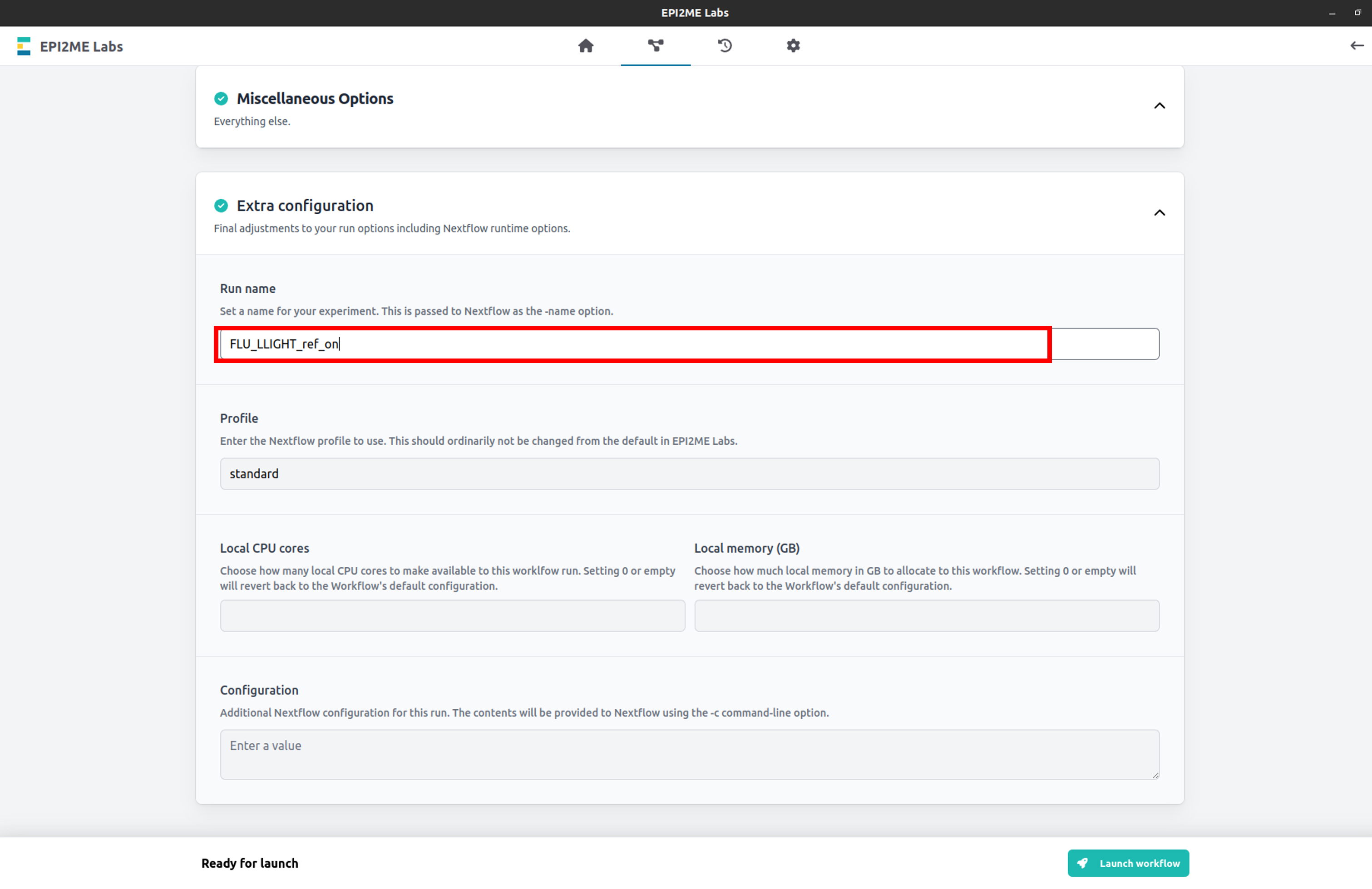
Click Launch workflow at the bottom of the page to begin your analysis.
-
Completed analysis and result files
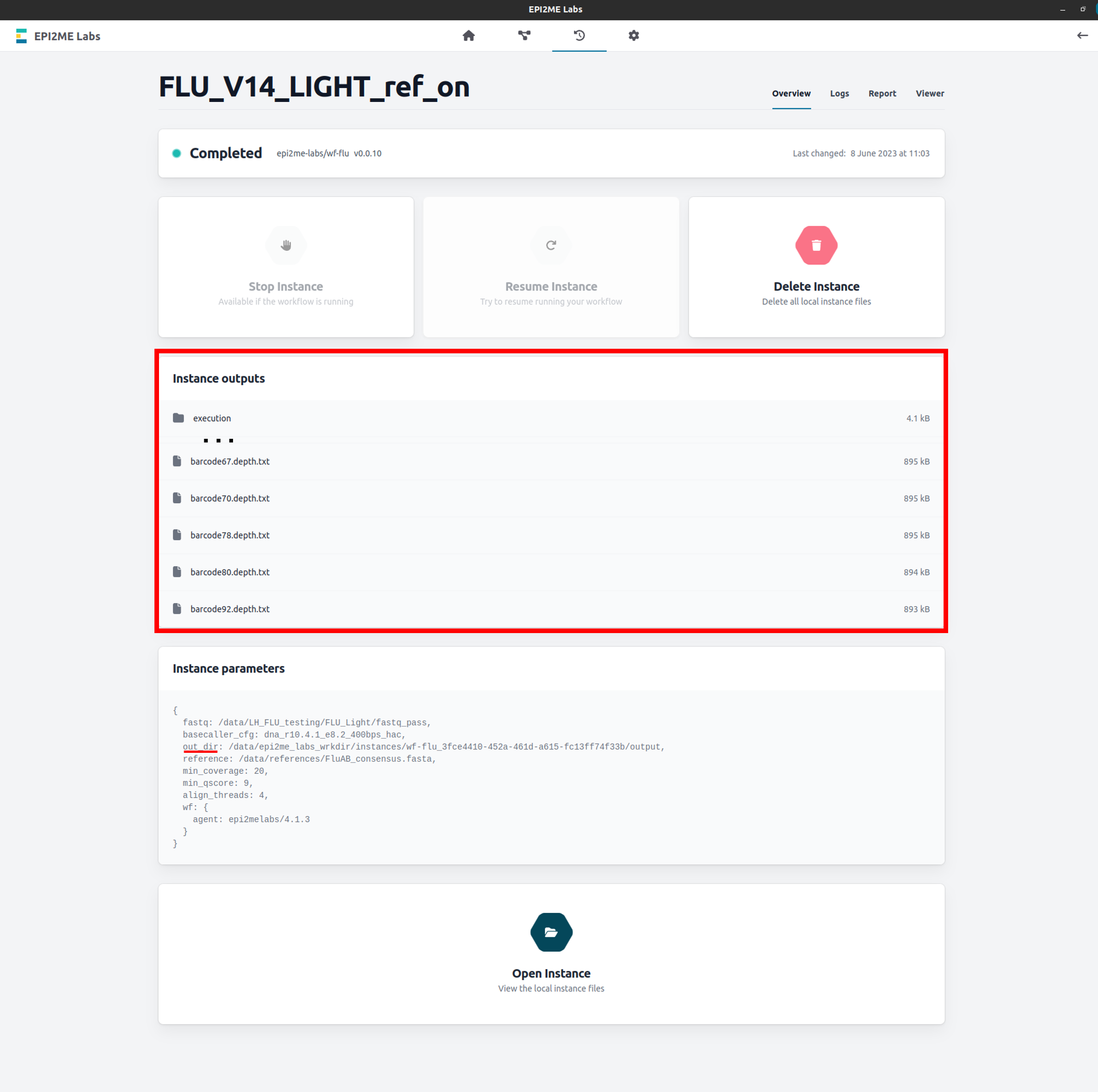
The wf-flu analysis outputs will be written to the Working Directory folder specified in the EPI2ME Labs Settings tab.
The location of this folder is specified in the wf-flu run Instance parameters preceeded byout_dir.However, these files can also be accessed directly in the EPI2ME Labs application from the completed analysis page for your run:
-
Housekeeping and disk usage
The "Working Directory" can be specified in the EPI2ME Labs "Settings" tab and defines where the workflow intermediate files and outputs are stored.
This folder will accumulate a significant number of files that correspond to raw BAM files, other larger intermediates and analysis results files. We recommend this folder to be routinely cleared.






