-
Post-basecalling analysis
We recommend performing downstream analysis using EPI2ME which facilitates bioinformatic analyses by allowing users to run Nextflow workflows in a desktop application. EPI2ME maintains a collection of bioinformatic workflows which are curated and actively maintained by experts in long-read sequence analysis.
Further information about the available EPI2ME workflows can be found here, along with the Quick Start Guide to start your first bioinformatic workflow.
For the analysis of amplicon sequences, we recommend using the wf-amplicon workflow which requires Nextflow and Docker or Singularity to be installed before running the workflow.
For installation instructions please click here.
-
Open the EPI2ME app using the desktop shortcut.
-
Navigate to the workflow downloads page. Click on the wf-amplicon workflow to download and confirm to install.
-
Navigate to the Workflows tab and click on wf-amplicon.
-
Click on "Run this workflow" to open the launch wizard.
-
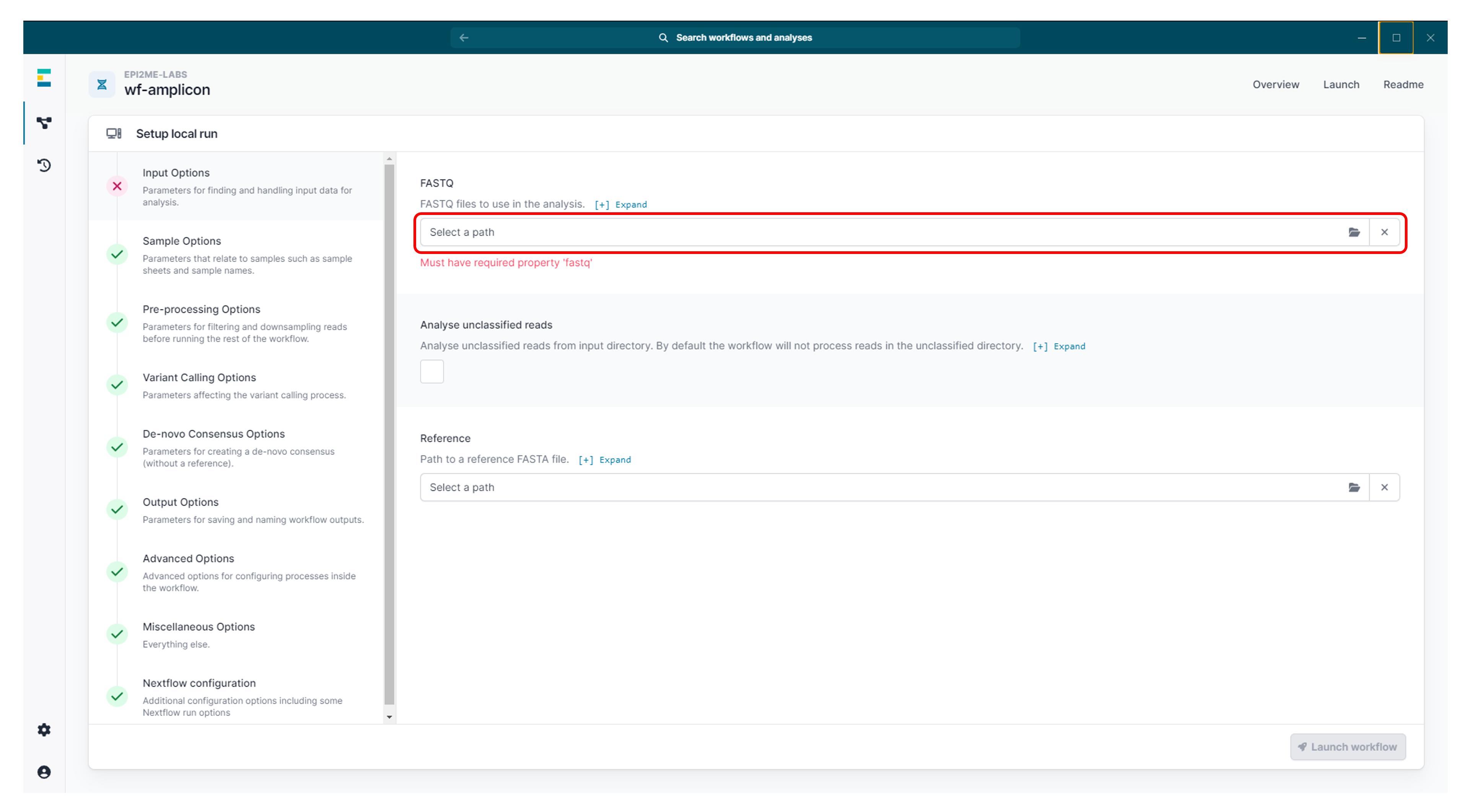
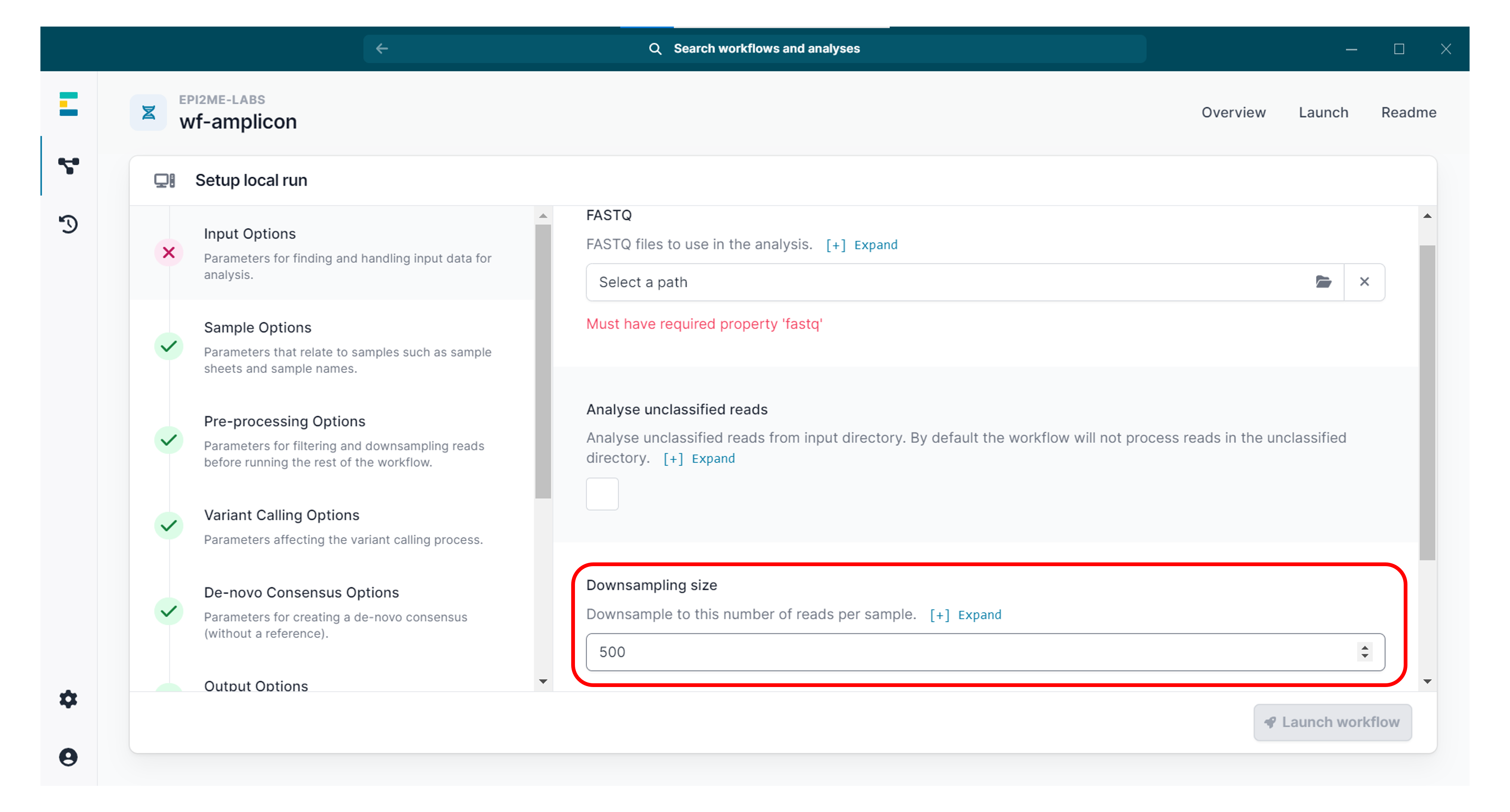
Set up your run by selecting your sequencing data in the "Input Options".
-
To speed up the analysis, you can set "Downsampling size" to 500.
Unless your amplicons are very long (>5 kb), this should provide sufficient coverage.
-
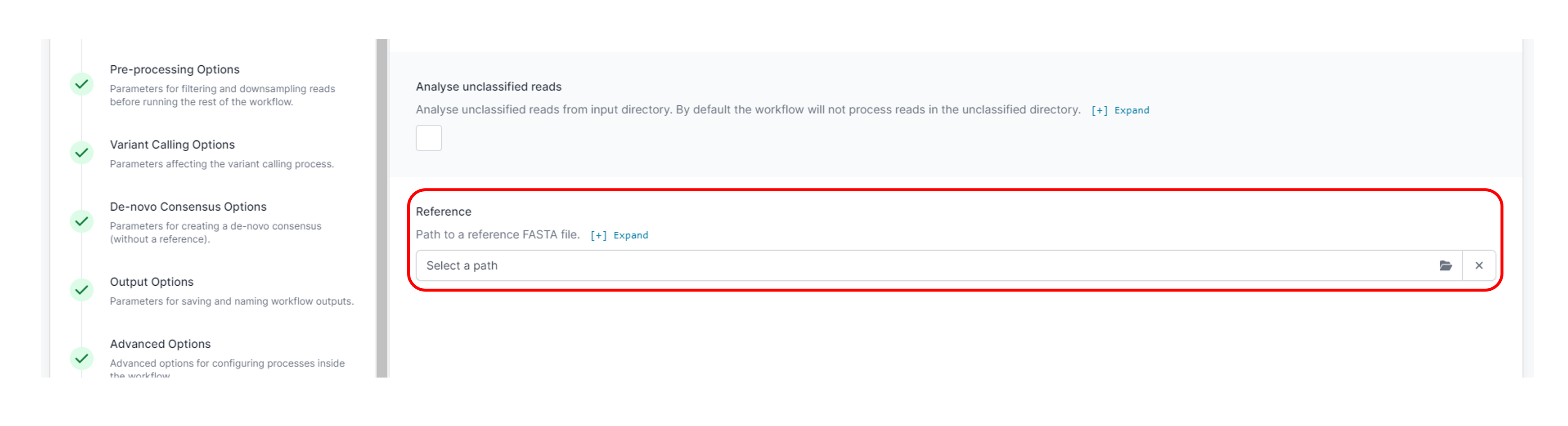
Optional actionThe amplicon workflow can be run with a sequence reference file if required.
Set up the reference FASTA by uploading the file in the following location:
-
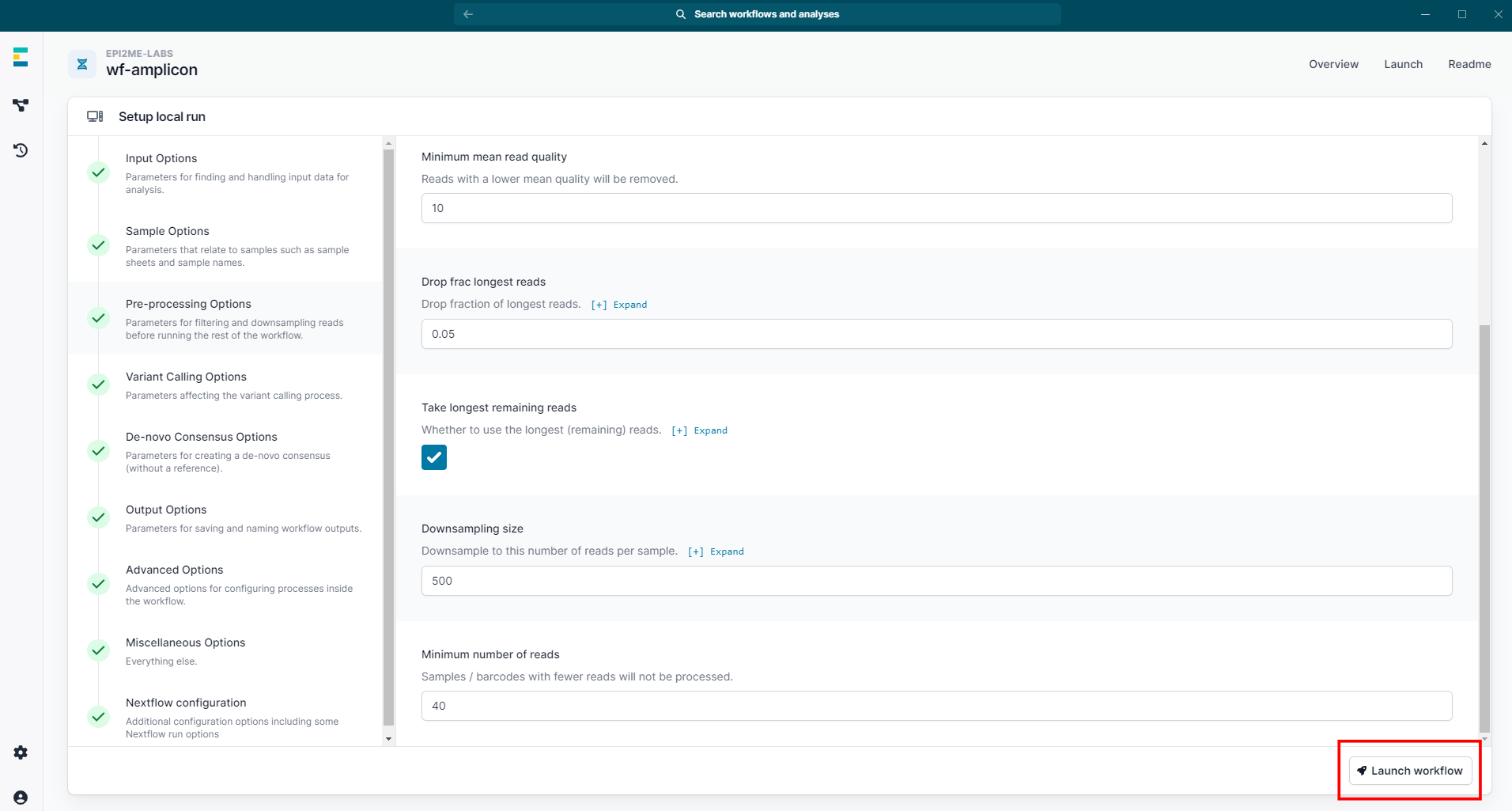
For the remaining parameter options we recommend keeping the default settings.
-
Click "Launch workflow".
Ensure all parameter options have green ticks.
-
Once the workflow finishes, a report will be produced.
-
Amplicon workflow report
The primary outputs of the workflow include:
· an interactive HTML report with tables and plots detailing the results.
· FASTQ files (one per barcode) with the de-novo consensus sequence and per-base consensus qualities (as calculated by Medaka).
· BAM files (one per barcode) of input reads re-aligned against the consensus.Example reports:
· When a reference file has been uploaded, reads are aligned to the reference (containing the expected sequence for each amplicon) for variant calling. An example of a sample variant calling report can be viewed here.
· When no reference file has been uploaded, the amplicon’s consensus sequence is generated de novo. An example of sample de novo consensus report can be viewed here.Report contents
The report consists of several sections. The introduction section gives a brief overview of key results for the individual samples analysed, while the preprocessing section illustrates the number of reads removed during downsampling / filtering. It also contains read length and quality histograms as well as a plot showing base yield vs. read length.
The remaining sections of the report depend on the mode in which the workflow was run. In variant calling mode, they summarise the mapping and variant calling stages of the workflow, showing the depth of coverage of aligned reads and providing further details on the variants called. In de novo consensus mode, results of the draft consensus QC stage and re-alignment of input reads against the consensus are described. Additionally, a depth of coverage plot of the re-aligned reads along the consensus is shown.






