-
Introduction to the Nanopore-only Microbial Isolate Sequencing Solution (NO-MISS) protocol
This end to end protocol describes our Nanopore-Only Microbial Isolate Sequencing Solution (NO-MISS): a flexible approach allowing sequencing of 4 to 24 microbial isolate genomes per MinION Flow Cell, generating a minimum coverage of 50x per genome.
The 50X coverage threshold is sufficient for downstream analyses including: accurate assembly and plasmid resolution, AMR profiling, core genome (cg) and whole genome (wg) multi-locus sequence typing (MLST), and cg/wgSNP typing. You can analyse your sequencing data using EPI2ME, which provides a user-friendly bioinformatics workflow.
We provide multiple DNA extraction approaches, depending on requirements, and starting organism (bacteria, fungi/yeast). These are key in achieving reliable flow cell output and genome coverage. The sample specific extraction methods use NEB Monarch Genomic Purification Kit, while the universal method uses a bead-beating method and the Maxwell RCS PureFood Pathogen Kit.
The extracted gDNA is then tagmented and sequenced using our Rapid Barcoding Kit (SQK-RBK114.24 or SQK-RBK114.96). Up to 24 samples per sequencing experiment for bacterial isolates (up to 7 Mb genomes) and up to 8 samples for fungi/yeast isolates can be processed to achieve the 50x coverage threshold. Use a minimum of four barcodes per run to maintain performance.
Detailed instructions for setting up the sequencing run on MinKNOW and downstream analysis are also included for a complete end-to-end protocol. We recommend sequencing up to 72 hours and generating at least 50x coverage per sample (approx. 0.5 Gb per barcode, assuming 5 Mb genome).We recommend updating MinKNOW to the latest version prior to starting a sequencing run. The basecalling model v4.3 found in Dorado 0.5.0 onwards provides improved accuracy for bacterial DNA and is included in MinKNOW release v24.02 or newer.
For more information on updating MinKNOW, please refer to our MinKNOW protocol.
End-to-end workflow overview
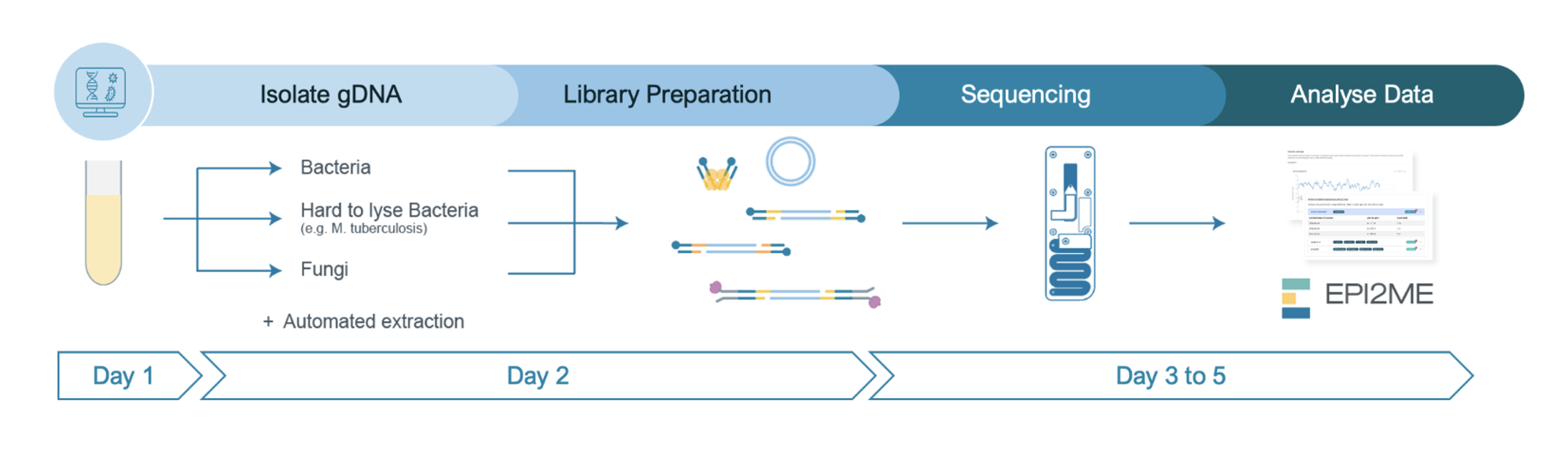
Steps in the sequencing workflow:
Prepare for your experiment
You will need to:
- Extract your DNA, and check quantity and purity. The quality checks performed during the protocol are essential in ensuring experimental success.
- Ensure you have your sequencing kit, the correct equipment and third-party reagents.
- Download the software for acquiring and analysing your data.
- Check your flow cell to ensure it has enough pores for a good sequencing run.
Sample preparation
Using the relevant gDNA extraction method, you will need to lyse your cells, extract your gDNA, and quantify the DNA:
- Universal bead-beating method:
- Universal bead-beating gDNA extraction:
- For high throughput requirements and universal applications.
- Manual column-based methods:
- Bacteria gDNA extraction:
- For bacteria, such as Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Serratia marcescens, Enterococcus faecalis, and Bacillus subtilis or staphylococci such as Staphylococcus aureus, and Staphylococcus epidermidis.
- Hard to lyse organisms gDNA extraction:
- For bacteria, such as Mycobacterium tuberculosis.
- Fungi gDNA extraction:
- For fungi/yeast, such as Candida albicans, Candida tropicalis, and Candida parapsilosis.
Library preparation
The table below is an overview of the steps required in the library preparation, including timings and stopping points.
Library preparation step Process Time Stop option DNA barcoding Tagmentation of the DNA using the Rapid Barcoding Kit V14 15 minutes 4°C overnight Sample pooling and clean-up Pooling of barcoded libraries and AMPure XP Bead clean-up 25 minutes 4°C overnight Adapter ligation Attach the sequencing adapters to the DNA ends 5 minutes We strongly recommend sequencing your library as soon as it is adapted Priming and loading the flow cell Prime the flow cell and load the prepared library for sequencing 5 minutes 
Sequencing and analysis
You will need to:- Start a sequencing run using the MinKNOW software, which will collect raw data to basecall and demultiplex the barcoded reads.
- Perform downstream analysis uing the isolate mode of the wf-bacterial-genomes workflow in EPI2ME.

