-
Post-basecalling analysis
We recommend performing downstream analysis using EPI2ME which facilitates bioinformatic analyses by allowing users to run Nextflow workflows in a desktop application. EPI2ME maintains a collection of bioinformatic workflows which are curated and actively maintained by experts in long-read sequence analysis.
Further information about the available EPI2ME workflows are available here, along with the Quick Start Guide to start your first bioinformatic workflow.
For the accurate and reliable assembly or alignment of bacterial or fungal isolate genomes generated from the included protocols, we recommend using the wf-bacterial-genomes workflow. At its core, the workflow is an efficient assembly pipeline which also polishes genomes using Medaka.
Whilst running the workflow using the default parameters will produce high quality genome assemblies, using the ‘Isolates’ mode will perform additional analyses designed to increase genome quality and aid genome interrogation for common pathogens in the clinical and food safety fields. ‘Isolates’ analyses includes MLST(7-gene), species confirmation and AMR prediction in addition to sample-specific reports.
Note: You can also run this workflow through command line. However, we only recommend this option for experienced users. For more information, please visit the wf-bacterial-genomes page on GitHub.
-
Open the EPI2ME app using the desktop shortcut.
-
On the landing page, open the workflow tab on the left-hand sidebar.
-
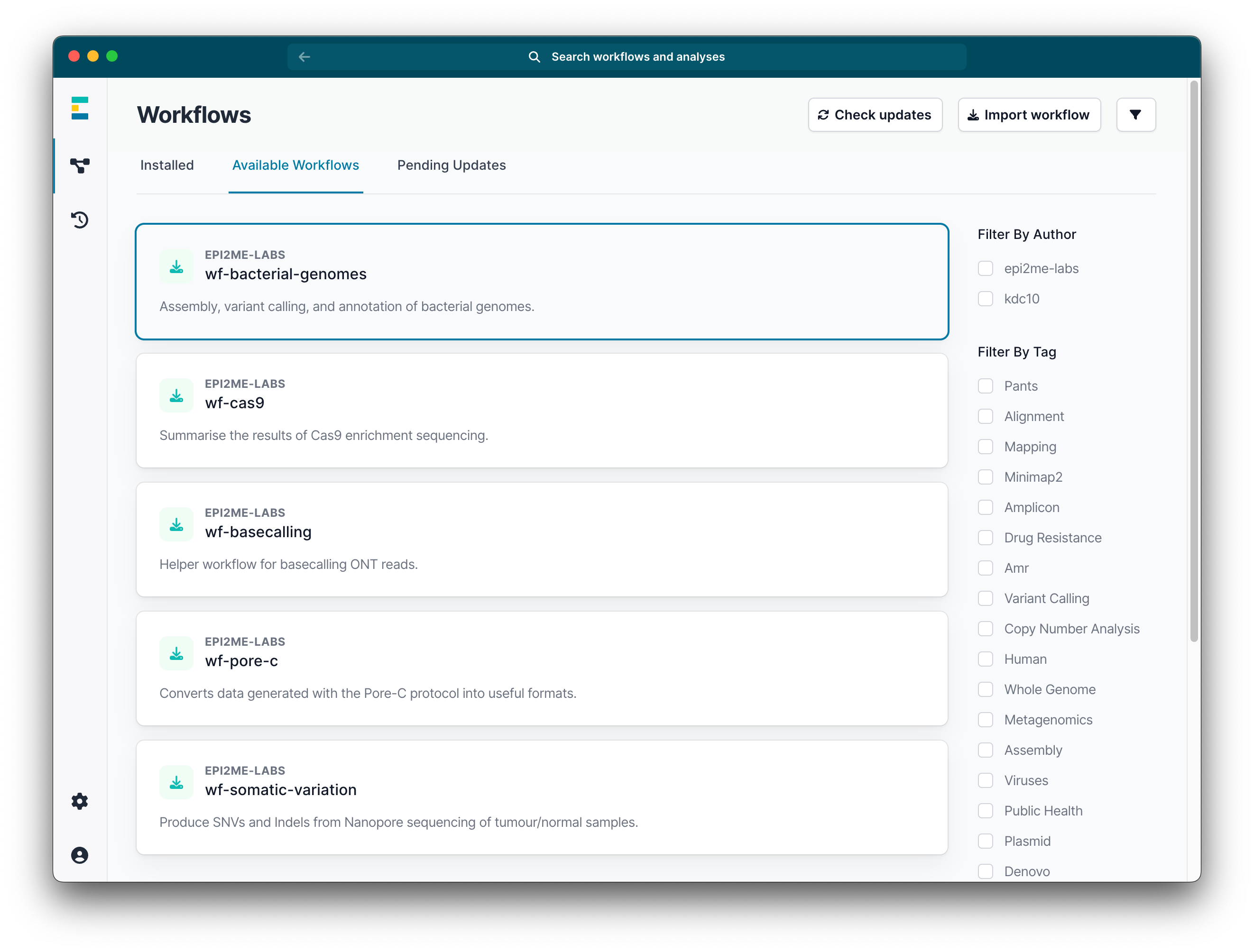
Navigate to the Available workflows tab and click on wf-bacterial-genomes option.
-
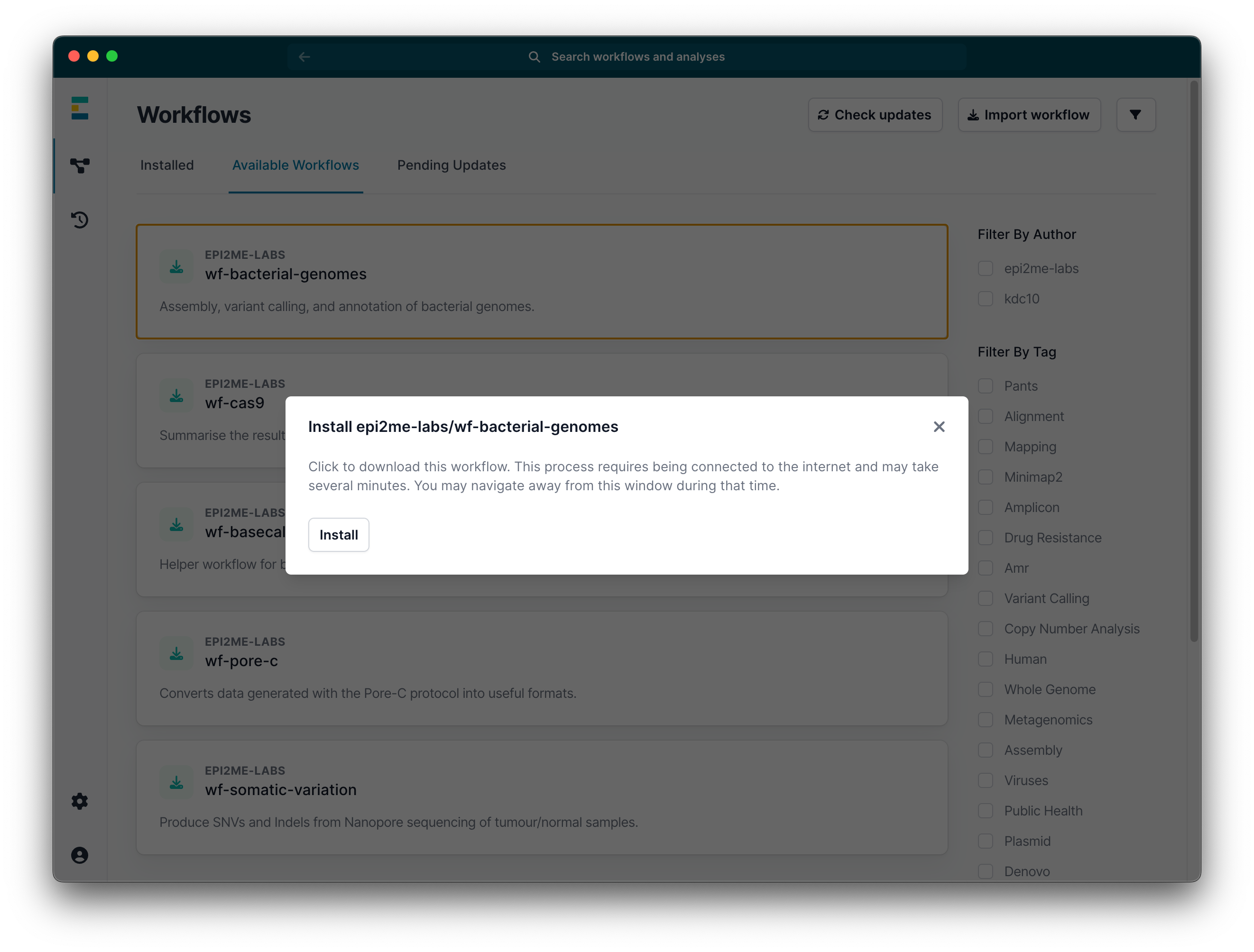
Click install.
-
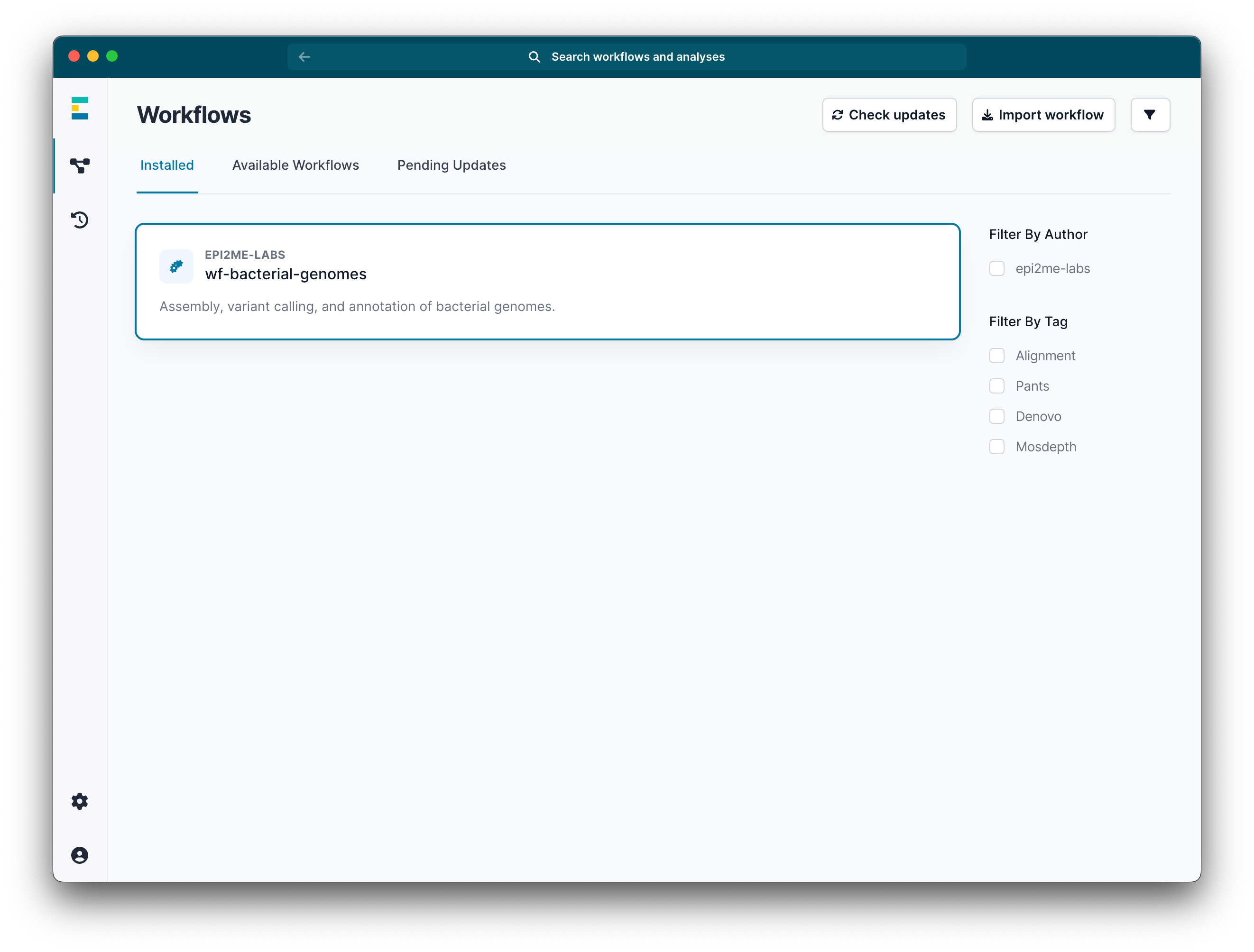
Navigate to the Installed tab and click on the installed wf-bacterial-genomes workflow.
-
Optional actionIf the workflow was already installed, check for updates by clicking 'Update workflow'.
We recommend running the latest version of our workflows for the best results.
-
Click on Run this workflow to open the launch wizard.
-
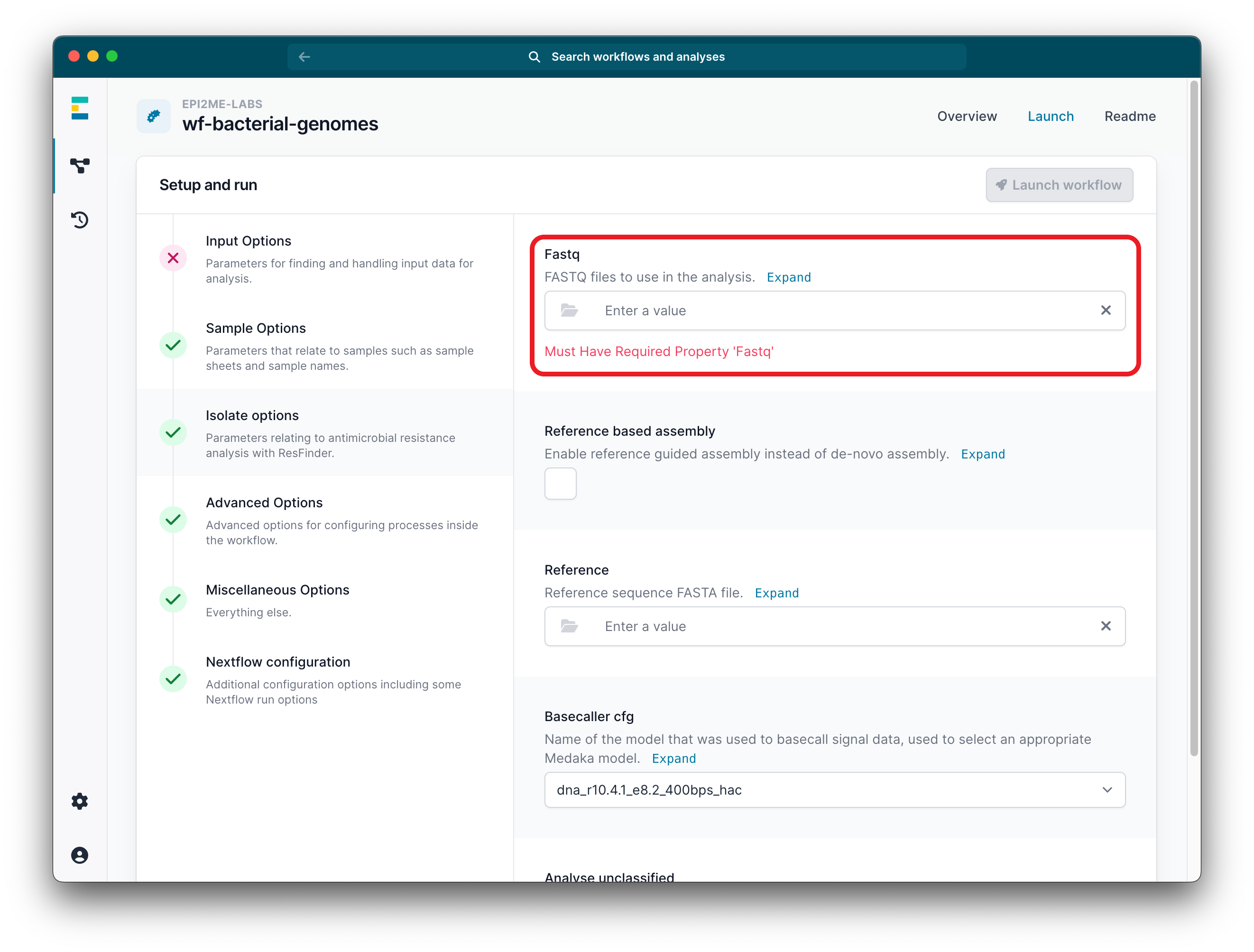
Set up your run by uploading your FASTQ file in the Input Options. We recommend keeping the default settings for the other parameter options.
-
To enable the isolates mode, tick the isolates checkbox.
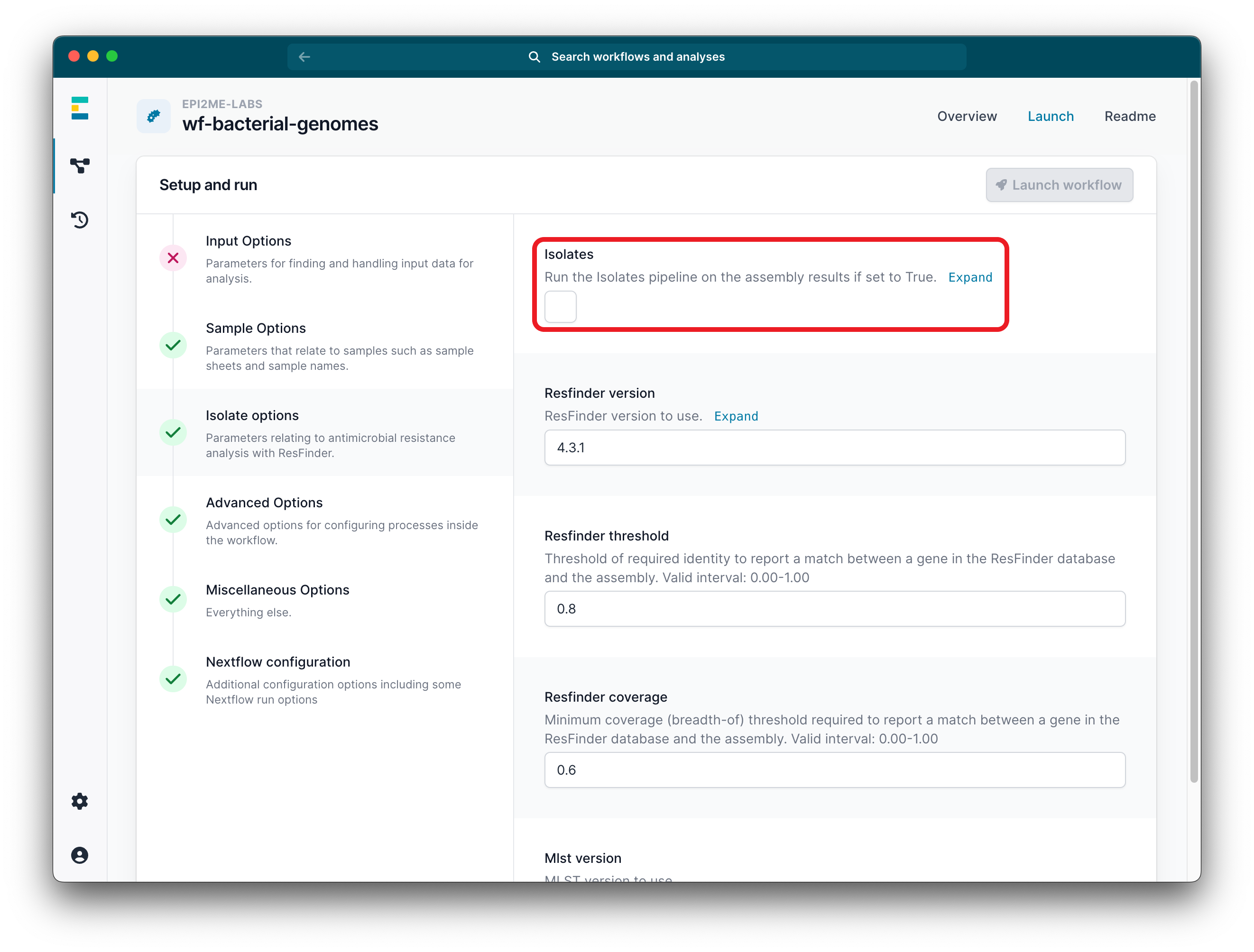
-
Navigate to the 'Nextflow configuration' tab to assign a 'Run name' to your analysis as an identifier.
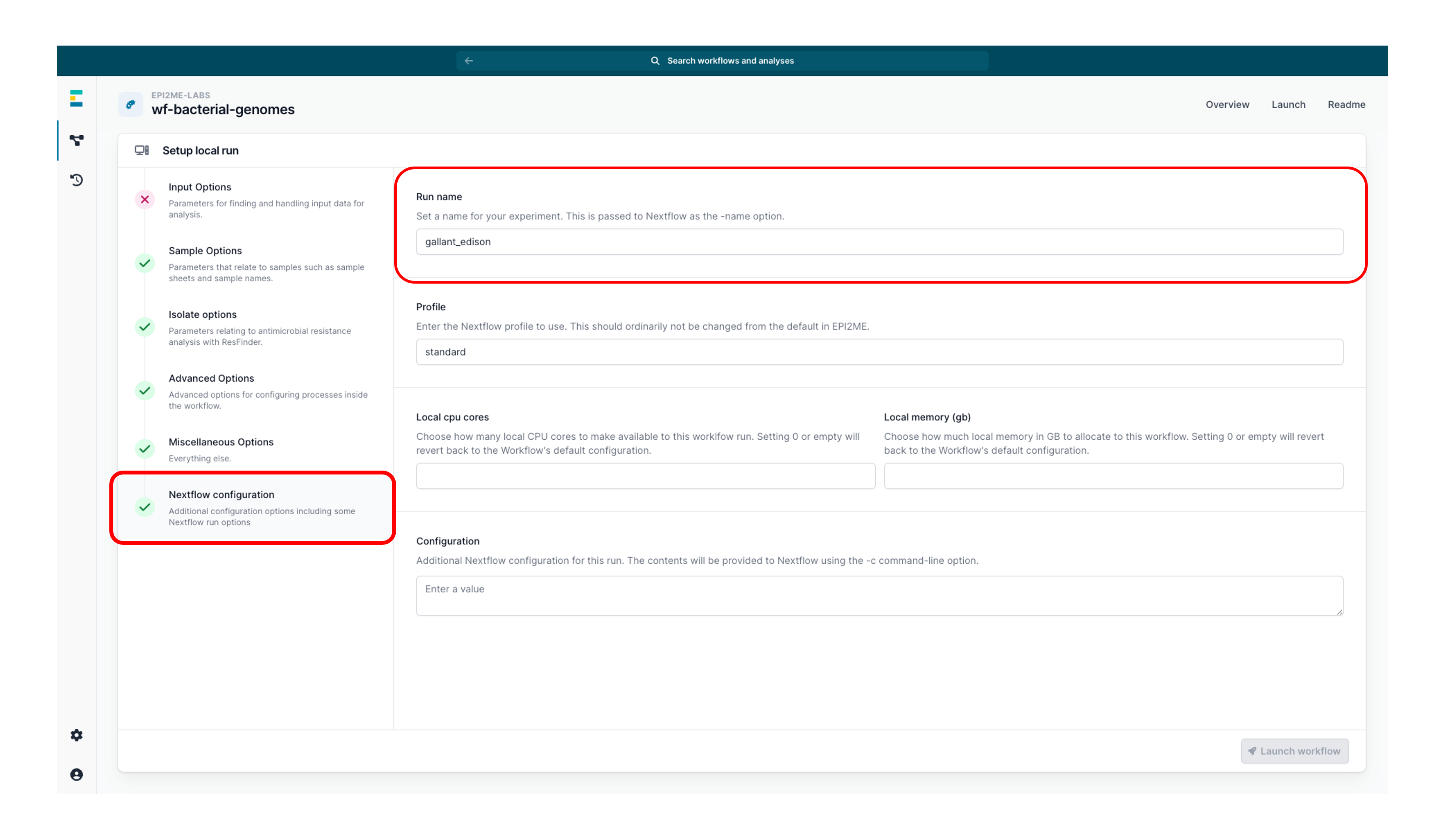
-
Click Launch workflow.
Ensure all parameter options have green ticks.
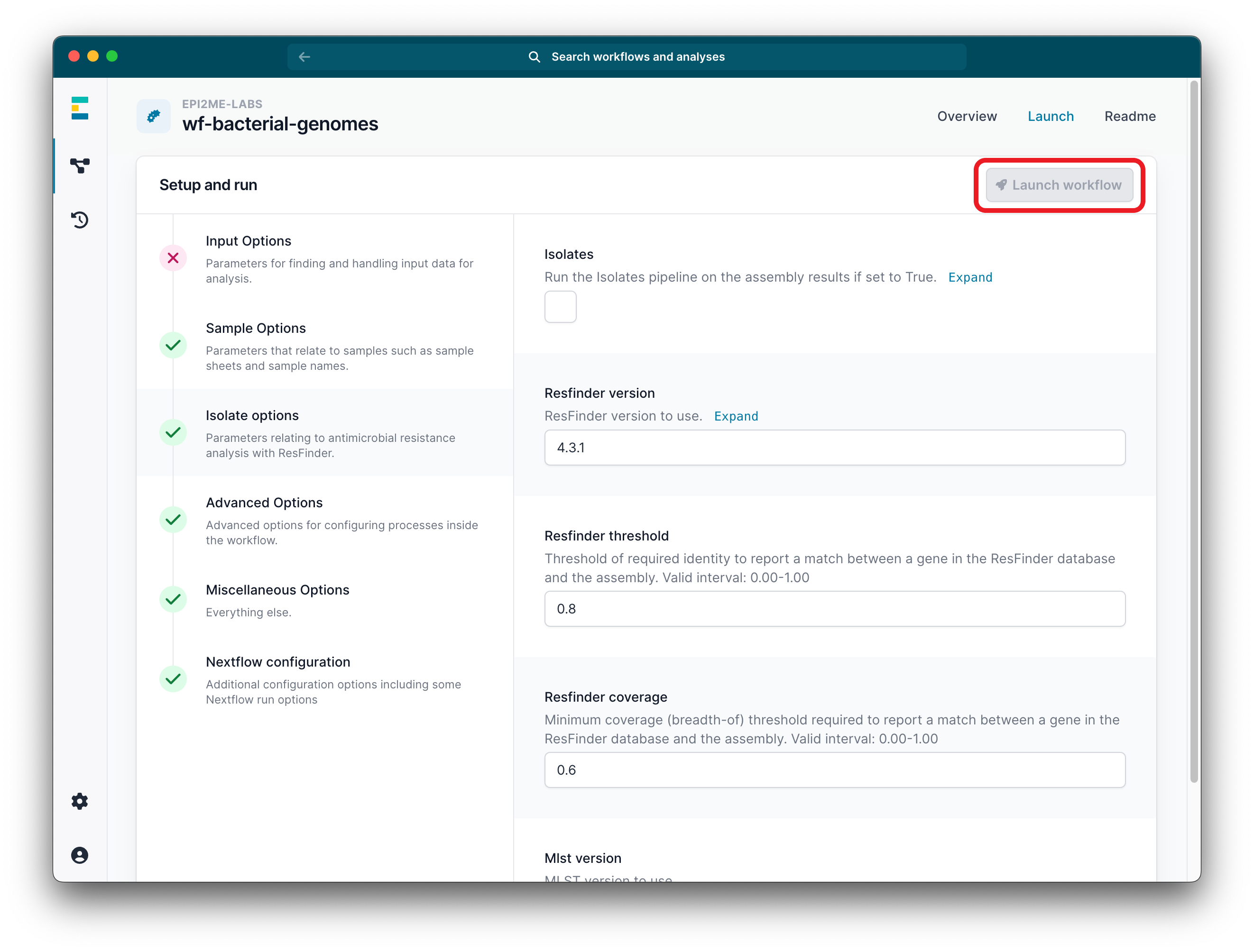
-
Once the workflow finishes, a report will be produced.










