-
Adaptive sampling inputs: creating a .bed file
After considering factors like % of the genome to enrich for, read lengths and response time, the next step is to collate the input files needed for an adaptive sampling experiment. These are:
- A genomic reference: this must be a FASTA or a pre-calculated minimap2 index
- Optionally, a .bed file with the genomic coordinates of the target regions
Note that if you do not input a .bed file, then the entire FASTA/minimap2 index file will be used for enrichment or depletion.
.bed files can be acquired in several ways.
Note: We recommend saving your alignment files in a folder with the prefix
/dataor the default location MinKNOW saves your reads after a sequencing run, to avoid issues when performing adaptive sampling in MinKNOW:e.g.
Windows: C:\data\
Mac: /Library/MinKNOW/data/
Ubuntu: /var/lib/MinKNOW/data/ -
If you are targeting an exome
The EPI2ME Labs analysis suite includes a "Curating Adaptive Sampling input files for MinKNOW" tutorial. The tutorial allows users to prepare and download the necessary files to perform an adaptive sampling experiment selecting for reads that span genes, transcripts, exons etc. stored within ensembl. The workflow outputs:
- A reference genome file
- The source .gtf file from which target regions were produced
- The .bed file containing target regions to provide to MinKNOW
To use EPI2ME Labs, refer to the Curating Adaptive Sampling input files for MinKNOW workflow.
-
If you are targeting other genomic regions
You will also need a .bed file and the reference sequence. Reference sequences can be downloaded from ensembl, UCSC Genome Browser, or other databases. For more information about .bed files and their required fields, see BED File Format - Definition and supported options.
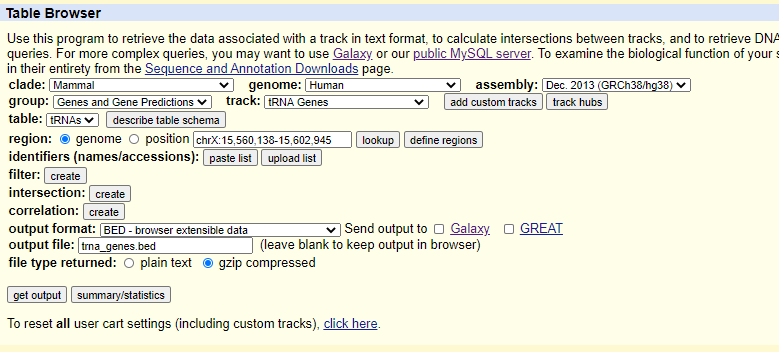
.bed files can be downloaded from the UCSC Table Browser. For example, to select human tRNA genes only:
- Select the following in the dropdown menus: a. clade: Mammal b. genome: Human c. assembly: Dec_2013(GRCh38/hg38) d. group: Genes and Gene Predictions e. track: tRNA Genes
- Select BED - browser extensible data in the output format.
- Click get output
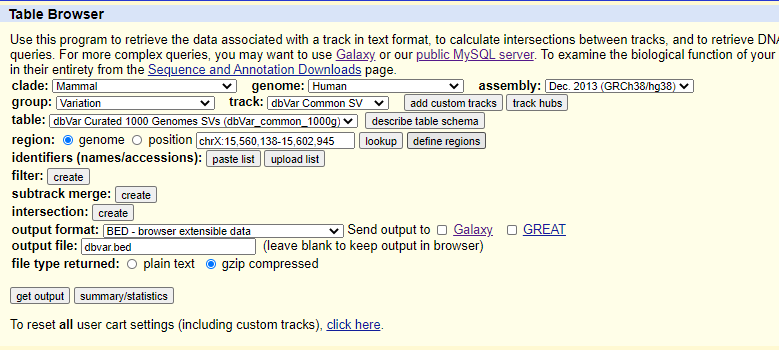
As another example, to select common structural variants in the human genome:
- Select the following in the dropdown menus: a. clade: Mammal b. genome: Human c. assembly: Dec_2013(GRCh38/hg38) d. group: Variation e. track: dbVar_Common_SV
- Select BED - browser extensible data in the output format.
- Click get output
-
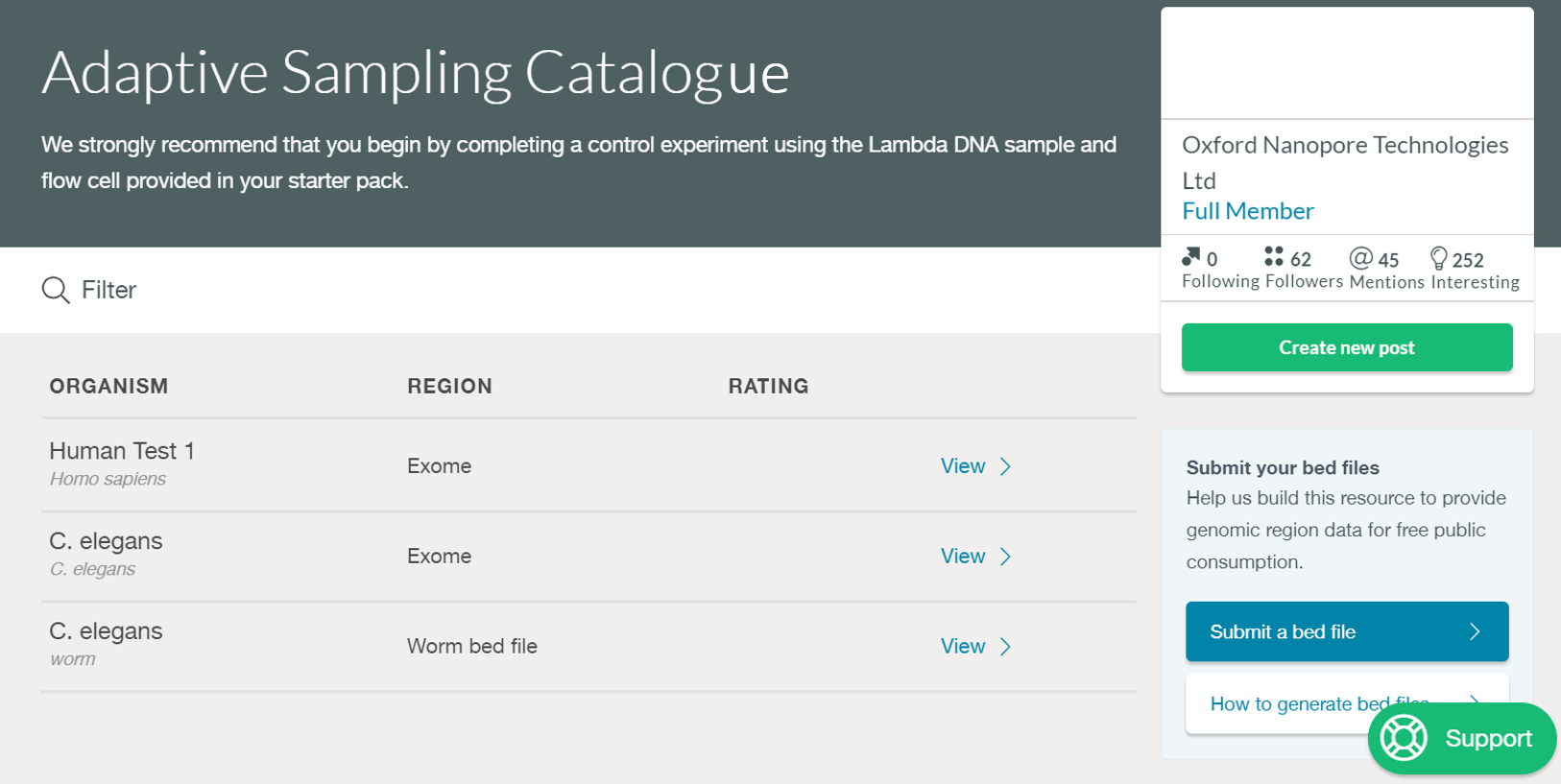
Sharing .bed files on the Nanopore Community
To browse adaptive sampling .bed files submitted by other Community members, or to submit your own, please visit the Adaptive Sampling Catalogue. Instructions are provided for how to add a .bed file to the catalogue.
-
Starting an adaptive sampling experiment in MinKNOW
Instructions for setting up an adaptive sampling run are provided in the MinKNOW protocol. During an enrichment experiment, adaptive sampling will reject all sequences that are not present in the reference/.bed file, whilst in a depletion experiment, only sequences that are present in the reference/.bed file will be rejected. Currently, adaptive sampling is enabled on MinION Mk1C, GridION and PromethION (for more details, see the "Release caveats" section below).
With adaptive sampling, barcode balancing is available as a beta feature which allows users to preferentially sequence under-represented barcodes in their samples to balance the read data across the barcodes based on the reference file provided. Please note, with increasing the number of reads sequenced for these barcodes, the overall data output for all reads may be reduced.
-
Output files
The files that MinKNOW outputs during a sequencing experiment are described in the MinKNOW protocol.
For adaptive sampling experiments, there is an additional CSV file named
adaptive_sampling.csvthat is saved inother_reportsin the run folder, which can be used for troubleshooting.The file has the following fields:
Field Description Example value batch_time The epoch time in seconds when the batch was processed 1595496307.7494152 read_number The order of the read within the channel 14 channel The channel on the flow cell that the read passed through 512 num_samples The number of samples in the read 4000 read_id The id of the read d605d893-44a3-4648-a5c3-50928267678f sequence_length The number of bases in the read. For stop_receiving reads, this is the number of bases recorded until the decision was made to continue sequencing the rest of the read. 324 decision Which decision was taken enrich: unblock, stop_receiving, unblock_hit_outside_bed
deplete: unblock, stop_receiving, stop_receiving_hit_outside_bed
An "unblock" decision means that adaptive sampling decided to reject the read.


