-
Post-basecalling analysis
We recommend performing downstream analysis using EPI2ME Labs which facilitates bioinformatic analyses by allowing users to run Nextflow workflows in a desktop application. EPI2ME Labs maintains a collection of bioinformatic workflows which are curated and actively maintained by experts in long-read sequence analysis.
Further information about the available EPI2ME Labs workflows are available here, along with the Quick Start Guide to start your first bioinformatic workflow.
For the mapping of AAV sequences for quality control and validation, we recommend using the wf-aav-qc workflow which requires Nextflow, Java, and Docker to be installed before running the workflow.
A run report will be produced and includes multiple plots to enable easy assessment of an AAV vector, including a contamination graph, truncations graph, transgene expression read coverage and genome type frequency graph.
-
Open the EPI2ME app using the desktop shortcut.
-
Navigate to the available workflows tab and click on the wf-aav-qc workflow to download and click install.
-
Navigate to the installed tab and click on wf-aav-qc.
-
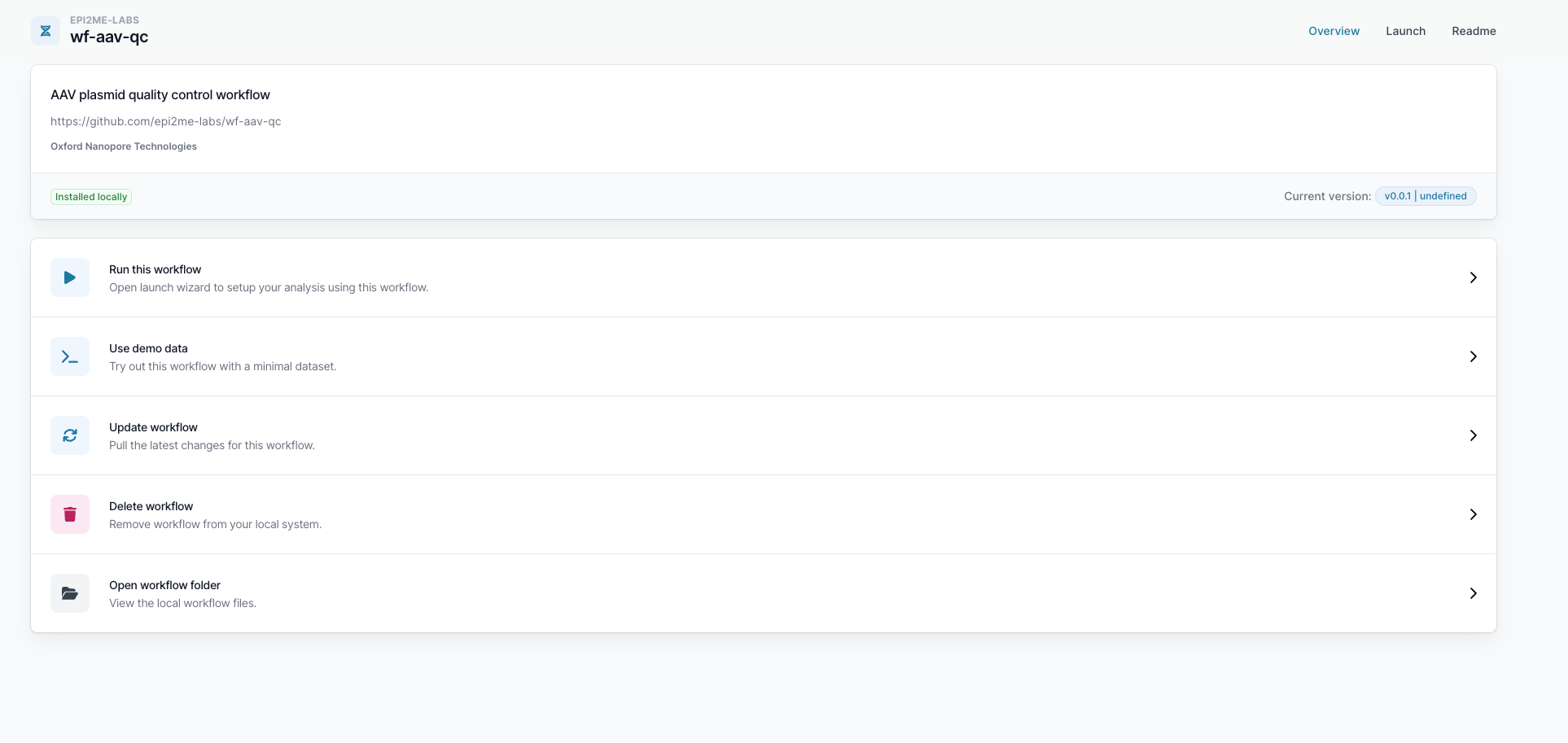
Click "Run this workflow" to open the launch wizard.
-
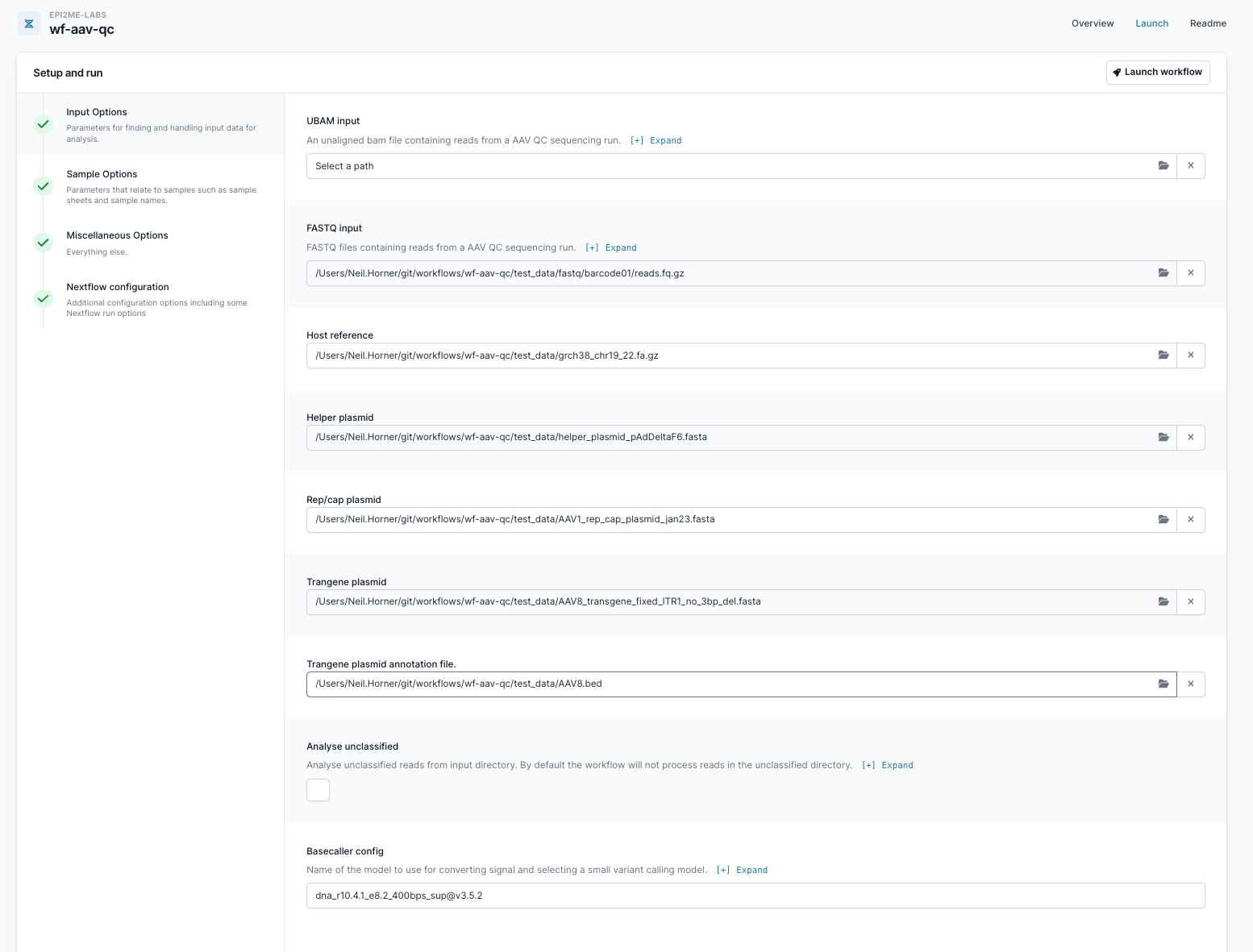
Set up your run by uploading your data files including the FASTQ input and host reference in the "Input Options". Fill in the basecaller configuration used to basecall your data and the other parameters can be kept to their default settings.
-
Click "Launch workflow" in the top right corner.
Ensure all parameters options have green ticks.
-
Once the workflow finishes, a report will be produced.





