-
Recommended pipeline analysis
The wf-artic is a bioinformatics workflow for the analysis of ARTIC sequencing data prepared using the Midnight protocol. The bioinformatics workflow is orchestrated by the Nextflow software. Nextflow is a publicly available and open-source project that enables the execution of scientific workflows in a scalable and reproducible way. The software is natively supported on the GridION device and can be simply installed on most Linux computers and servers. The installation is outlined later in the document.
The Midnight analysis uses the ARTIC bioinformatics workflow.
Demultiplexed sequence reads are processed using the ARTIC FieldBioinformatics software that has been subtly modified for the analysis of FASTQ sequences prepared using Oxford Nanopore rapid sequencing kits. The other modification to the ARTIC workflow is the use of a primer scheme that defines the sequencing primers used by the Midnight protocol and their genomic locations on the SARS-CoV-2 genome.
The wf-artic workflow includes other analytical steps that include cladistic analysis using Nextclade and strain assignment using Pangolin. The data facets included in the report are parameterised and additional information such as plots of depth-of-coverage across the reference genome is optional.
The complete source for wf-artic is linked and the Nextflow software will download the scripts and logic flow from this location.
On GridION devices, the wf-artic workflow will start automatically after sequencing. However, on other devices, this will have to be started manually as outlined further on this page under 'Running a Midnight analysis'.
-
Software set up and installation
The wf-artic workflow requires the Nextflow and Docker software to have been installed. The EPI2ME quickstart guide provides instructions for the installation of these requirements for GridION, PromethION and general Ubuntu Linux users and provides a little more introduction to the Nextflow software.
Automatic start on GridION:
To set up the Midnight analysis to start automatically after sequencing on GridION, select the Rapid Barcoding Kit 96 (SQK-RBK110.96) kit with the Midnight RT PCR Expansion (EXP-MRT001) pack on MinKNOW when setting up a sequencing run.
When the workflow has finished, the relevant analysis files will be available in the following output folder:
processing/artic/artic_DATE_TIME_67195e17Post-run analysis on GridION:
The Midnight analysis can also be started post-run on GridION:
1. On the start page, click 'Analysis'
2. Click 'Workflow'
3. From the dropdown menu, select 'post_processing/artic/artic'
4. Select your input folder with the sequencing data and the location for the output folder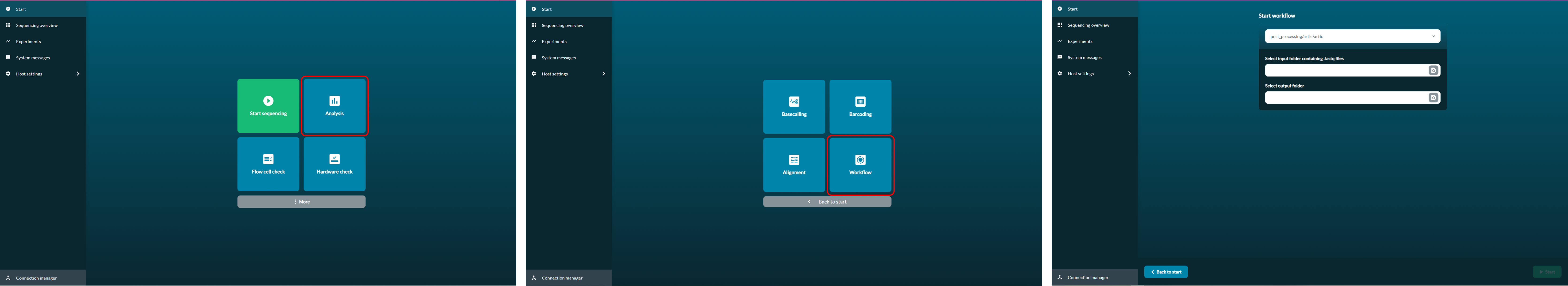
Using Linux command line:
The wf-artic workflow can be run from the Linux command line. The workflow can be installed or updated with the command:
$ nextflow pull epi2me-labs/wf-artic -
Demultiplexing of multiple barcoded samples
The wf-artic requires FASTQ format sequence data that has already been demultiplexed. Sequences can either be demultiplexed directly in the MinKNOW software or as a post-sequencing step by the guppy_barcoder software provided by the Guppy software.
The Midnight protocol uses a rapid barcoding kit; it is therefore important to note that the demultiplexing step must not require barcodes at both ends of the sequence.
The expected input for wf-artic is a folder of folders as shown below. Each of the barcode folders should contain the FASTQ sequence data and files may either be uncompressed or gzipped.
$ tree -d MidnightFastq/MidnightFastq/├── barcode01├── barcode02├── barcode03├── barcode04├── barcode05├── barcode06└── unclassified -
Running a Midnight analysis
The reference command for running a Midnight analysis is as follows. The parameters are explained further on in the document.
nextflow run epi2me-labs/wf-artic \--scheme_name SARS-CoV-2 \--scheme_version V1200 \--min_len 200 \--max_len 1100 \--out_dir PATH_TO_OUTPUT \--fastq PATH_TO_FASTQ_PASS \-work-dir PATH_TO_INTERMEDIATE_FILESType the command into you linux terminal and press enter.
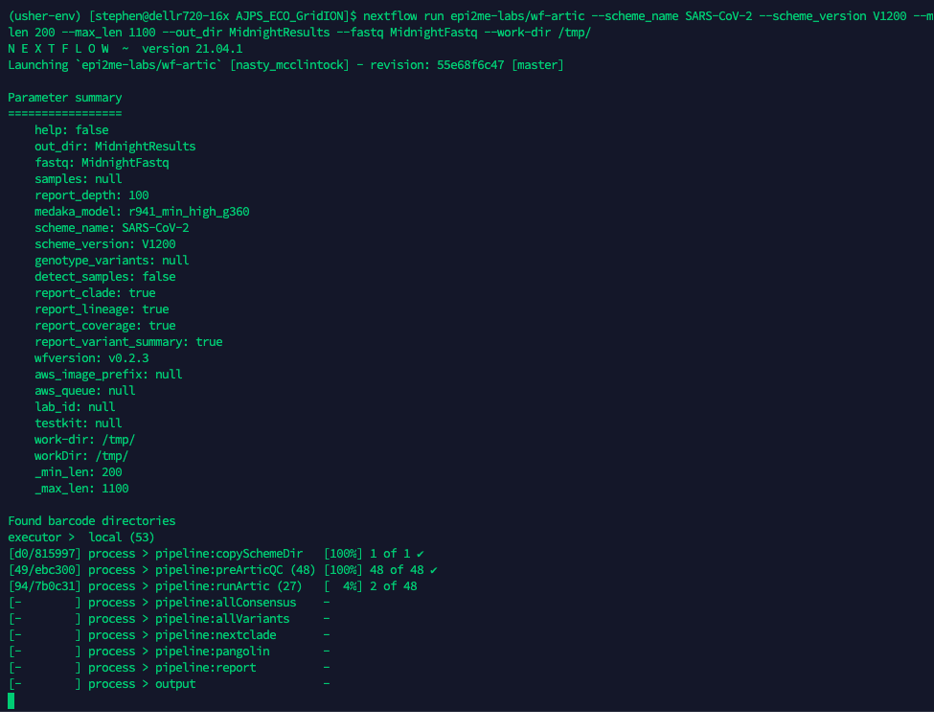
Nextflow will describe the analysis as it progresses; the figure above shows an example run from a 48-plex analysis. We can see which processes have completed and the processes that are still running and or queued.
-
Parameter definitions
nextflow run epi2me-labs/wf-artic
An instruction to use the Nextflow software to run a workflow, which is further explained here.--scheme_name SARS-CoV-2
An instruction for the ARTIC software to use the primer scheme that corresponds to the amplicons tiled across the whole SARS-CoV-2 genome.--scheme_version V1200
This defines the version of the ARTIC primers to use. The Midnight protocol uses the primer set refered to as V1200.–-min_len 200
This sets the minimum allowed sequence length as 200 nucleotides.–-max_len 1100
This sets the maximum allowed sequence length as 1100 nucleotides.--out_dir PATH_TO_OUTPUT
This instructs the Nextflow software where the results should be stored; please change PATH_TO_OUTPUT to the location on your computer where files should be stored.--fastq PATH_TO_FASTQ_PASS
This instructs Nextflow which sequences should be used in the analysis. Please changePATH_TO_FASTQ_PASSto an existingfastq_passfolder from a Midnight run.-work_dir PATH TO WORK DIRECTORY
Please note the single hyphen; this is a Nextflow parameter.
This defines where the intermediate files are stored. This folder may contain a significant amount of information; please see the section on housekeeping.
Other command line parameters
Other commands and options can be provided to the Nextflow command:
--samples
This describes a sample file that links barcode identifier with sample names. These sample names will be reported in the HTML format report and in the CSV file of genotypes. The sample file should be a comma-delimited file and must contain the column names barcode andsample_name.--help
This will display the help-file which describes the available parameters and other information on default values and their meanings.--medaka_model
This defines the model that should be used by the Medaka software for variant calling (and thus consensus preparation).
-
Result files
Results will be written to the location specified by the
--out_dirparameter. These output results include:all_consensus.fasta
A multi-FASTA format sequence file containing the consensus sequence for each of the samples investigated. This consensus sequence has been prepared for the whole SARS-CoV-2 genome, not just the spike protein region. The consensus sequence masks the non-spike regions and regions of low sequence coverage with N residues.all_variants.vcf.gz
A gzipped VCF file that describes all high-quality genetic variants called by medaka from the sequenced samples.all_variants.vcf.gz.tbi
An index file for the gzipped VCF file.consensus_status.txt
A tab delimited file that reports whether a consensus sequence has been successfully prepared for a sample, or not.wf-artic-report.html
A report summarising these data. This HTML format report also includes the output of the Nextclade software that can be used for a visual inspection of, for example, primer drop out or other qualitative consensus sequence aspects.
Other files are included in the
work-directory. This includes per sample VCF files of all genetic variants prior to filtering and other sequences. -
Housekeeping and disk usage
The nextflow parameter,
-work-dir, was introduced as a parameter to define where the workflow intermediate files are stored. This folder will accumulate a significant number of files that correspond to raw BAM files and other larger intermediates. We recommend this folder to be routinely cleared. -
Updating the wf-artic software
Updated versions of the wf-artic software may be released and an alert to the availability of newer workflow versions will be noted by the Nextflow software at run-time.
To update the software:
nextflow pull epi2me-labs/wf-articIt may be necessary to first delete the cached workflow files. This can be achieved with the command:
nextflow drop -f epi2me-labs/wf-artic

