-
Post-basecalling analysis
We recommend performing downstream analysis using EPI2ME Labs which facilitates bioinformatic analyses by allowing users to run Nextflow workflows in a desktop application. EPI2ME Labs maintains a collection of bioinformatic workflows which are curated and actively maintained by experts in long-read sequence analysis.
Further information about the available EPI2ME Labs workflows are available here, along with the Quick Start Guide to start your first bioinformatic workflow.
For the assembly of small plasmid sequences, we recommend using the wf-clone-validation workflow which requires Nextflow and Docker to be installed before running the workflow.
-
Open the EPI2ME app using the desktop shortcut.
-
Scroll down on the landing page and click on the wf-clone-validation workflow to download and confirm to install.
-
Navigate to the Workflows tab and click on wf-clone-validation.
-
Click on "Run this workflow" to open the launch wizard.
-
Set up your run by uploading your FASTQ file in the "Input Options". We recommend keeping the default settings for the other parameter options.
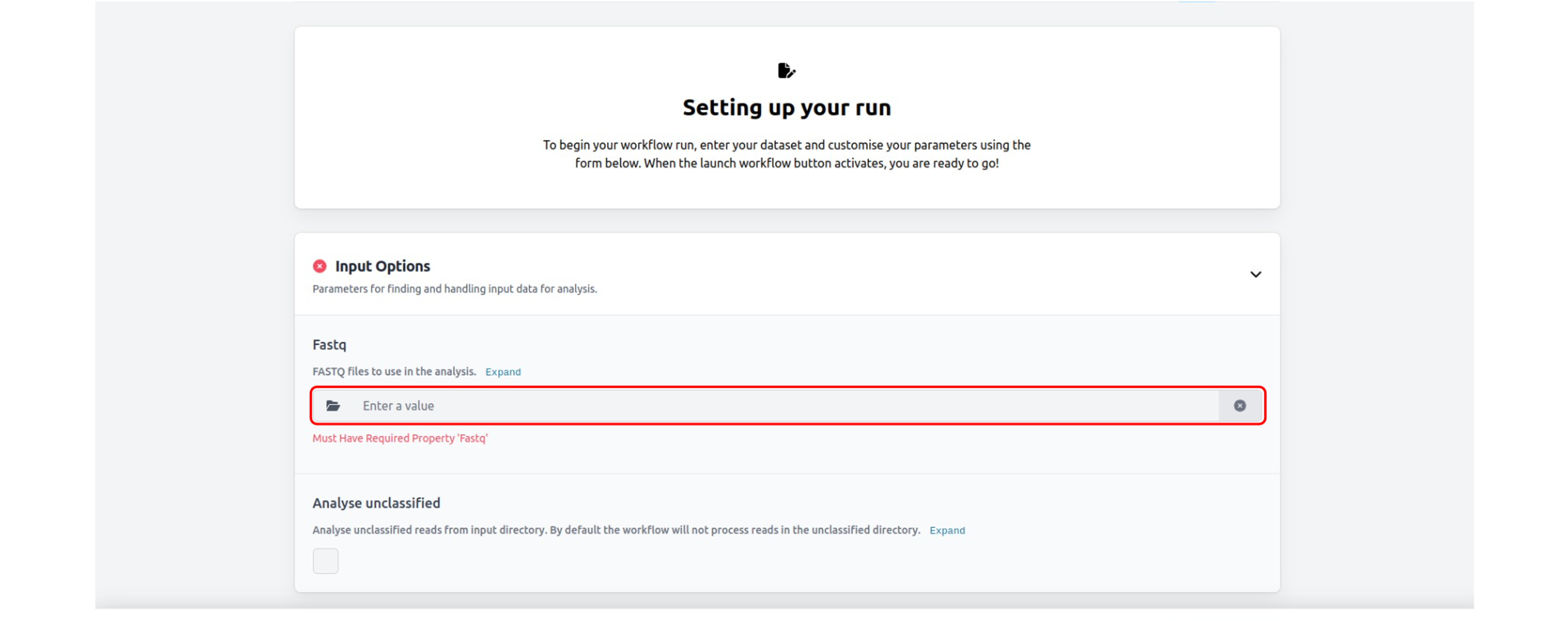
-
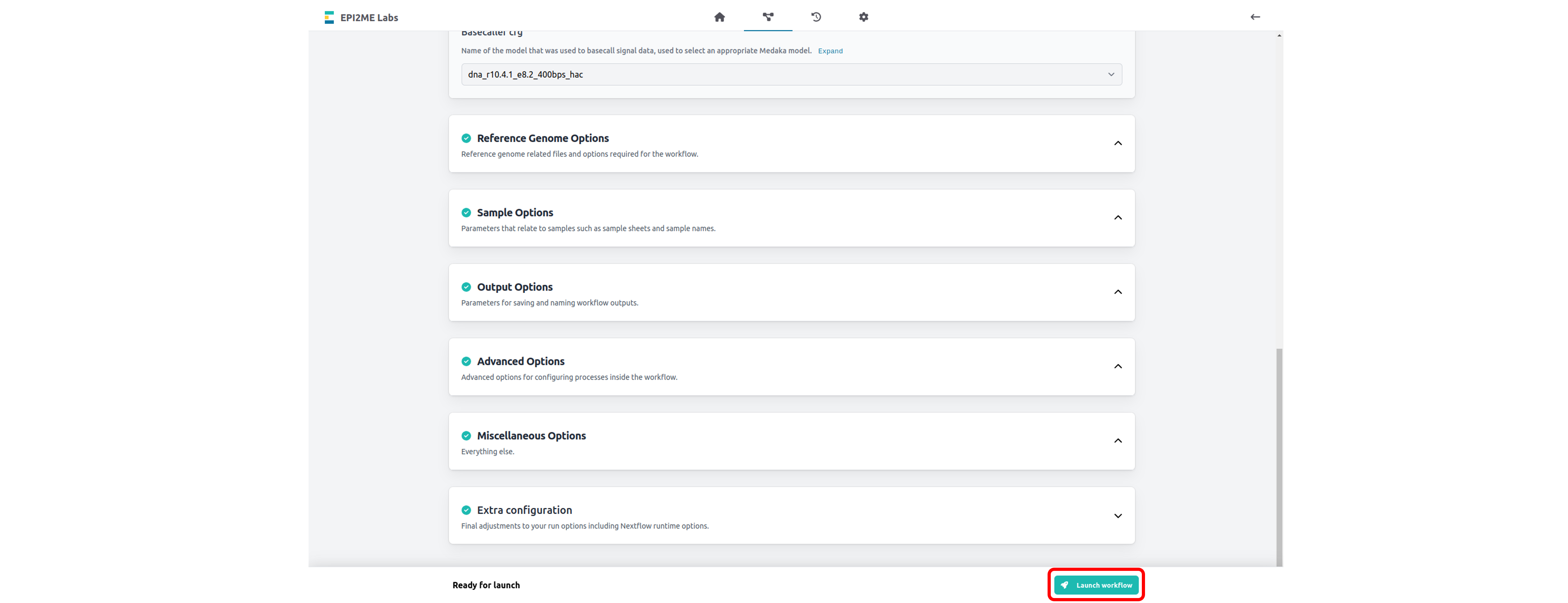
Click "Launch workflow".
Ensure all parameter options have green ticks.
-
Once the workflow finishes, a report will be produced.
-
Clone validation workflow report
A report is produced containing the results of the assembled plasmid sequences. The primary outputs of the workflow include:
- a consensus .fasta file for each sample
- a .csv showing the pass or fail status of each sample
- a feature table containing annotations for each of the samples
- an HTML report document detailing the primary findings of the workflow
A sample report can be viewed here.
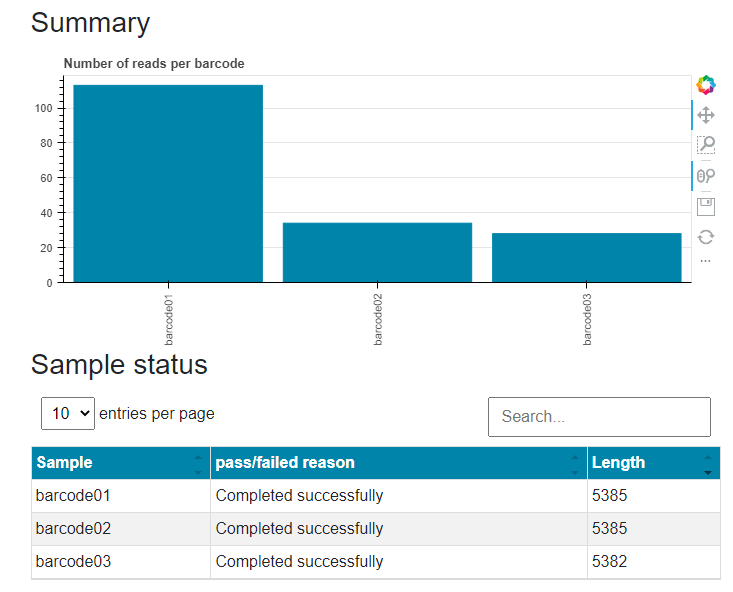
The summary graph shows the number of reads per barcode and the table shows the length of the consensus sequence for each barcode. These data may be used to identify the samples that may have dropped out of the sequence analysis due to insufficient sequence reads.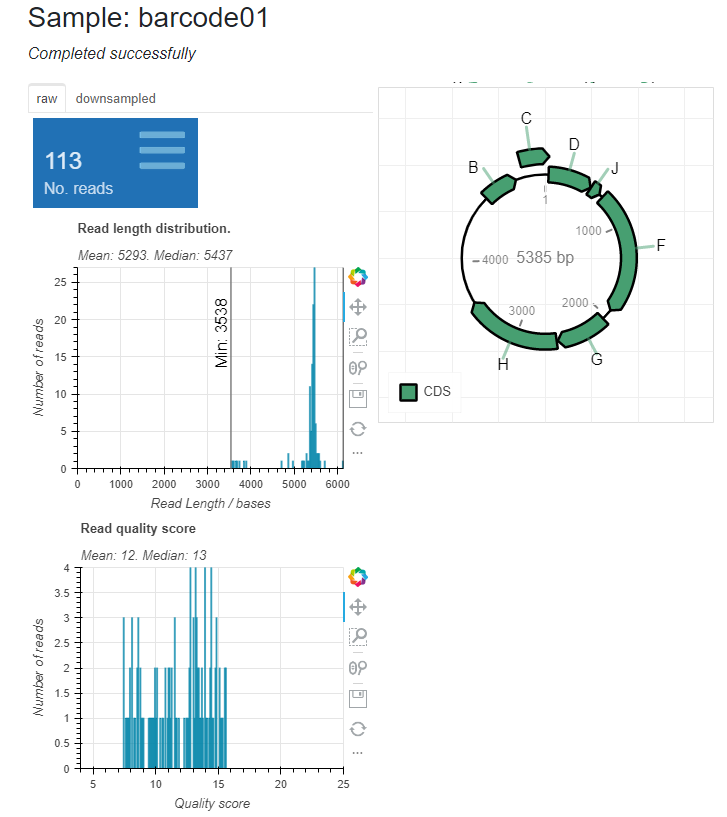
For each barcode, read length statistics and a pLannotate plot is presented to illustrate the polished plasmid consensus sequence. In the sample report, barcode01 has been assembled into a 5385 bp consensus sequence. The unfilled features on the plot are incomplete features.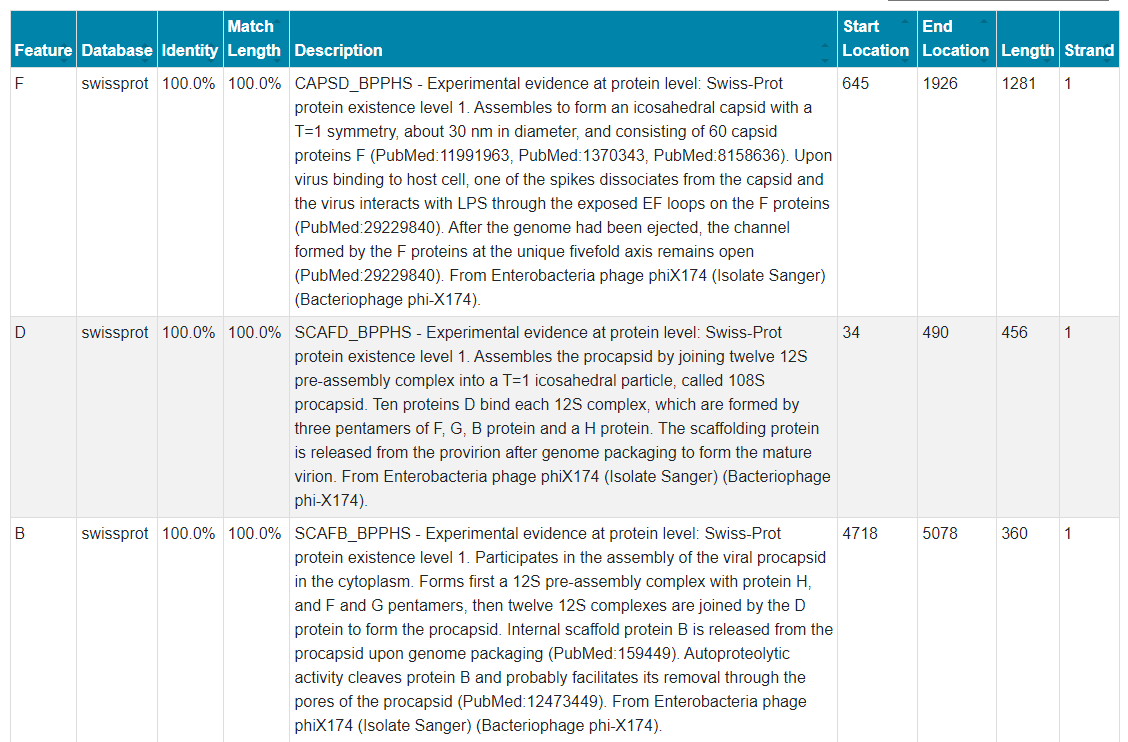
A feature table is also provided for each barcode to give descriptions of the annotated sequence which can be used to identify the precise location of the annotated features.








