-
Fast, High Accuracy and Super Accurate models and compatibilities
The MinKNOW basecallers offer three different basecalling models: a Fast model, a High accuracy (HAC) model, and Super accurate (SUP) model.
The Fast model is designed to keep up with data generation on Oxford Nanopore devices (MinION Mk1C, GridION, PromethION). The HAC model provides a higher raw read accuracy than the Fast model and is more computationally-intensive. The Super accurate model has an even higher raw read accuracy, and is even more intensive than the HAC model.
For more information about basecalling accuracy, see the Accuracy page on the Oxford Nanopore website.
A comparison of the speed of the models is provided in the table below:
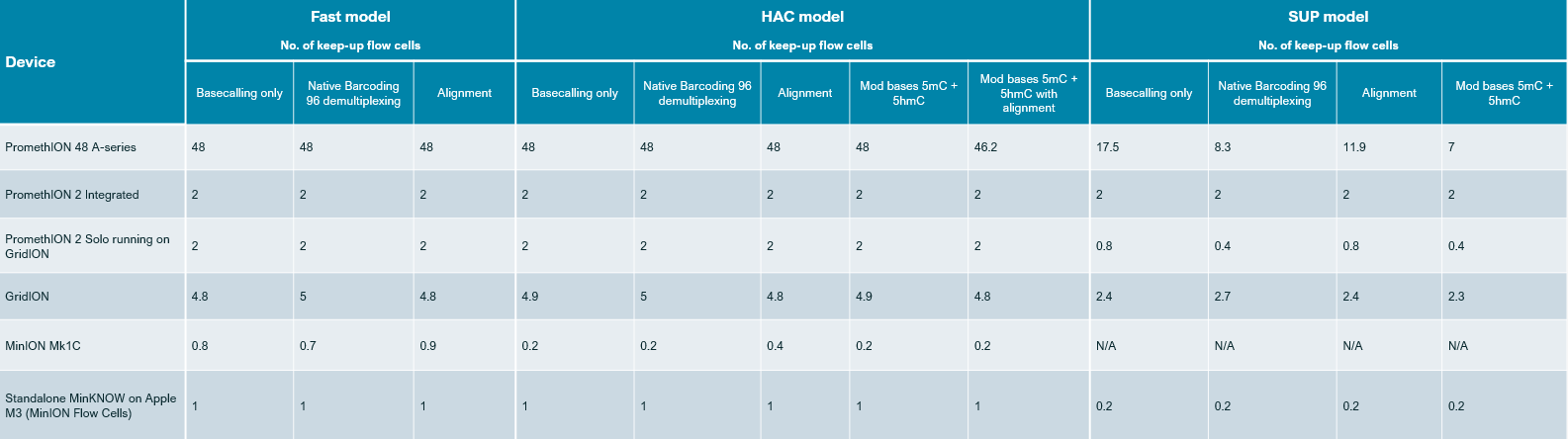
The number of keep-up flow cells assumes a 30 Gbase flow cell output in 72 hours for MinION and GridION, and 100 Gbase output in 72 hours for PromethION.
-
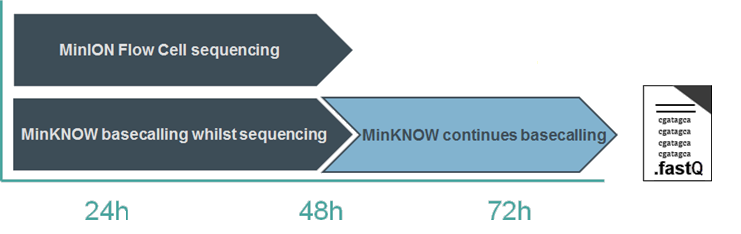
MinKNOW basecalling: keep-up vs catch-up
Basecalling with the Fast basecalling model can keep up with the speed of data acquisition on most nanopore platforms. High Accuracy basecalling keeps up on GridION, and with 18 flow cells on PromethION A-Series. When using the more computationally-intensive models, basecalling continues after the sequencing experiment has run to completion; any reads that have not been basecalled during the experiment will be queued and processed afterwards. This is known as “Catch-up mode”.
You therefore have two options: either to allow MinKNOW to continue in catch-up mode, or to stop the analysis and basecall the remaining reads at a later time, e.g. using stand-alone Dorado.
-
Calling modified bases
Base modifications, including 5mC, 5hmC, and 6mA for DNA and m6A for RNA, can be called from nanopore signal data. This requires the use of a designated basecalling model that is trained to identify base modifications. The simplest way to access these models is via MinKNOW on the device, or the standalone Dorado basecaller from GitHub. MinKNOW currently has models for 5mC + 5hmC (CG-context and all-context) and 6mA (all-context) for DNA, and a m6A model for RNA operating in a DRACH context. Standalone Dorado includes these models alongside other models, including 4mC + 5mC for DNA and pseudouridine for RNA. The basecalling software outputs modified base information in BAM files.
Several advanced options are also available for calling and analysing modified bases. Remora is a tool available on GitHub that provides the tools to prepare datasets, train modified base models and run simple inference. Another option is to use modkit (also available on GitHub) for post-processing base modifications after basecalling. Modkit creates summary counts of modified and unmodified bases in an extended bedMethyl format. bedMethyl files tabulate the counts of base modifications from every sequencing read over each reference genomic position.
If you wish to train your own all-context modified base calling models, we are now offering a limited developer release of the software tool Betta for the processing of “randomer” datasets. A randomer is a chemically synthesized oligonucleotide with a specific construct including a fixed width section of randomly inserted canonical bases. Betta provides a chemistry protocol and easy-to-use commands for generation and analysis of data from this construct design. The primary target of these pipelines is a Remora dataset for input into training a Remora modified base detection model. If you would like access to this tool, register your interest here.
-
Basecaller, consensus and variant caller model training
When developing basecalling, consensus, and variant-calling models using machine learning, Oxford Nanopore Technologies uses data from sequencing experiments. This data can be synthetic or derived from genomic sources. Model development is broken down into two broad categories: training (creating a model) and validation (showing that it works). The rest of this section will focus on training basecall models, although similar strategies apply to the other model types.
To train a DNA or RNA basecall model, sequencing experiments using a range of genomes are run to generate raw signal data (.pod5). This data is then prepared for training by selecting a representative subset of reads; basecalling them with Dorado; and aligning them to a ‘ground truth' reference. Once the data is prepared, the new basecall model is then trained using Bonito software, which applies machine-learning methods to fit a model to the training dataset. Various additional parameters can be set to configure the basecalling training appropriately for the sequencing condition, which are described in further detail in the Bonito documentation.
Typically, the training dataset contains raw signal data from pod5 files including samples of human, C. elegans, and ZymoBIOMICS Microbial Community Standard sequencing experiments. The data includes both PCR-amplified reads and native reads that can contain base modifications. A portion of the reads and/or genomic locations are reserved for validating the model and not included in the training dataset.
Once trained, the quality of the model is validated using reads covering genomic regions that were not included in the training dataset. Validation assesses the following parameters:
- Alignment accuracy
- % of strands that align to the reference
- Identifying strand edges and barcodes
- Specific test cases such as low complexity and homopolymer sequences
- Basecalling in and around methylation motifs
- De novo genome assembly quality
- Consensus accuracy (with and without trained polishing models)
- Short variant calling (SNPs and indels, with and without trained polishing models)
- Structural variants
If the validation meets the minimum criteria and the new model is an improvement on the currently-released models, it is then included in Oxford Nanopore's production software.

