-
Basecalling options
We offer two options for basecalling Kit 12 data with sequencing accuracies of 99% and above (Q20+):
- Simplex basecalling, where the template DNA strand passes through the nanopore and is basecalled.
- Duplex basecalling, where the complement strand is read immediately after the template strand and the consensus basecall for both strands leads to a further increase in accuracy.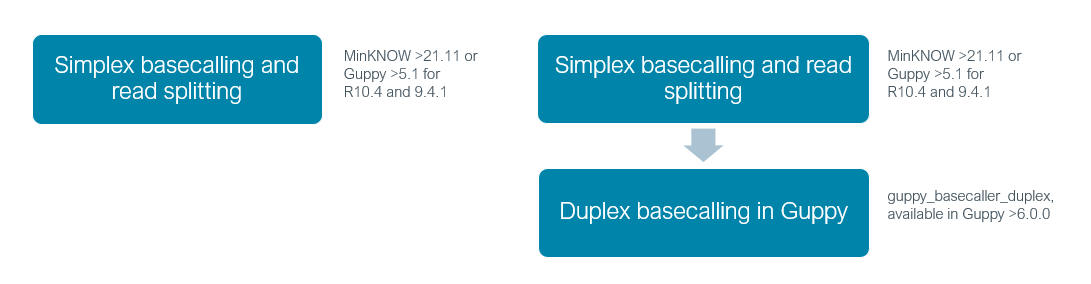
Each of these options is described in more detail in the "Basecalling Kit 12 data" subsections:
-
We recommend basecalling Kit 12 data in MinKNOW.
For optimal performance, we recommend using a GridION, PromethION, or a computer with:
- 64 bit Linux (Windows will be supported in future)
- Intel i7, i9, Xeon, or better processor
- At least 16 GB of RAM
- An NVIDIA GPU, at least RTX 2070 or better, with at least 16 GB of GPU memory
- At least 1 TB SSD
Make sure you are using the most recent version of MinKNOW.
-
Set the sequencing parameters as described in the MinKNOW protocol under "Starting a sequencing run", and choose Kit 12 kits (e.g. SQK-LSK112) under Kit selection.
-
Introduction to read splitting
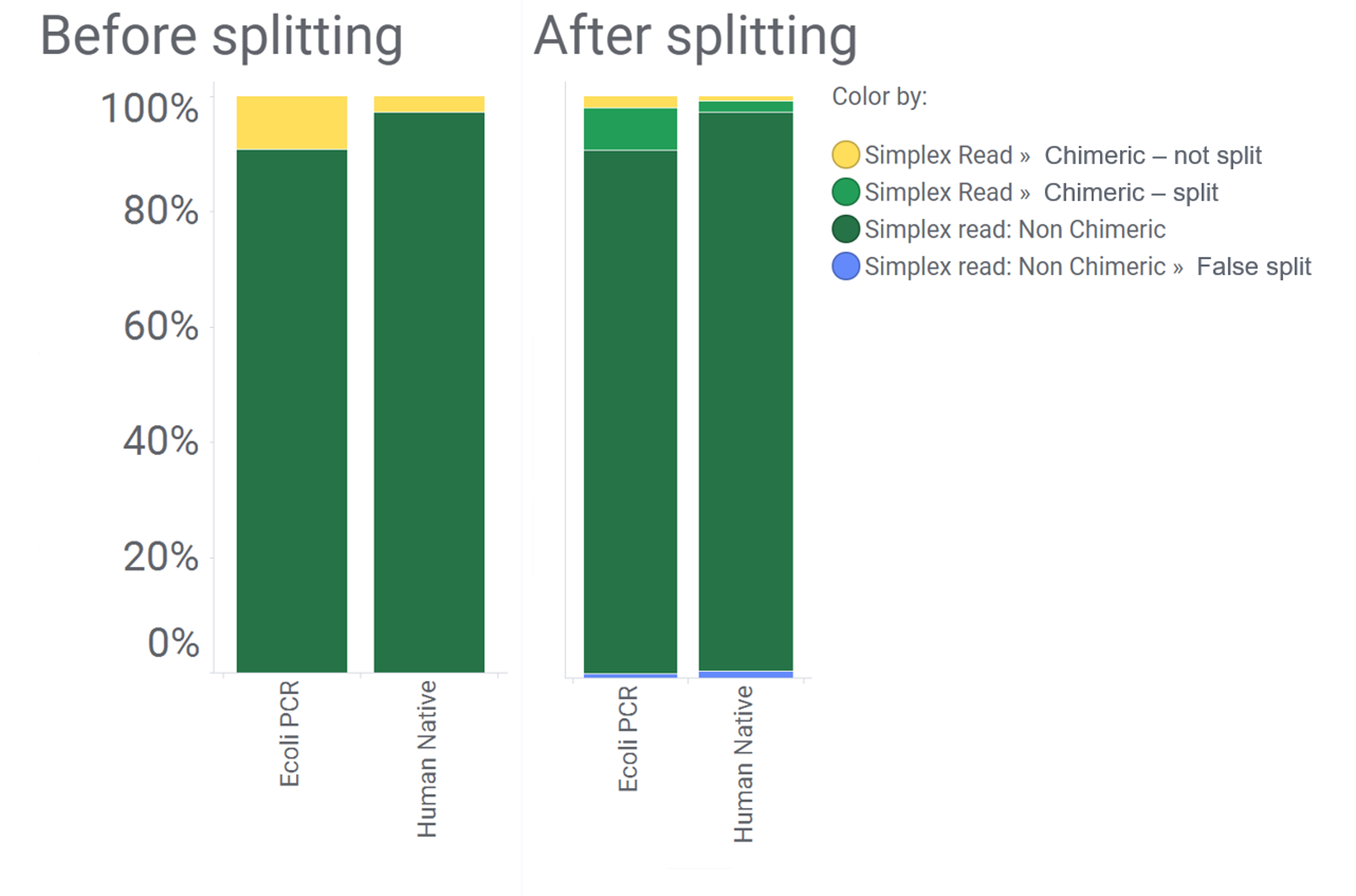
With increased follow-on rates of the Kit 12 chemistry (the rate of the complement strand entering the pore directly after the template strand has passed through), we have observed a higher rate of concatemerisation compared to the Ligation Sequencing Kit (SQK-LSK110). We are classifying these reads as 'informatic chimeras' as they are not physically joined during the library preparation process.
With SQK-LSK110, we typically observe <2% concatemerisation and at this rate, it typically does not affect downstream applications. With, for example SQK-LSK112, we have observed a rate as high as 10%. Both MinKNOW (v21.11 and higher) and stand-alone Guppy (v5.1 and higher) now offer the option of splitting these reads.
As you can see from the following example, the majority of the informatic chimeras (yellow) are removed after splitting for a human (native) and E. coli (PCR) sample.
It is important to note that the read splitting function is not designed to split reads that are incorrectly ligated together during sample preparation. While these make up a small percentage of reads, users should take care with ligation steps to follow the protocol carefully to reduce the chance of creating them.
-
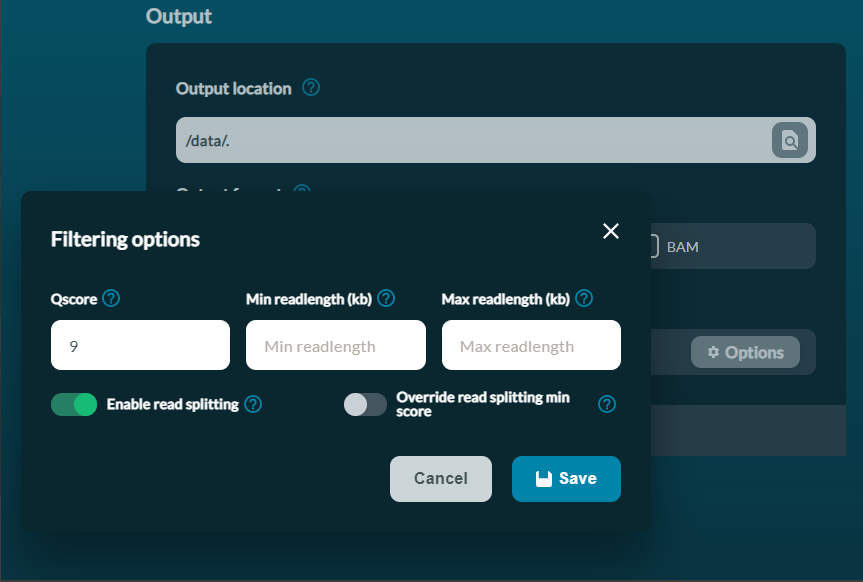
Enable read splitting in MinKNOW
When configuring your experiment, under Output - Filtering options, toggle "Enable read splitting" to ON.