-
Download (or prepare) the relevant portion of target genome plus flanking sequence in FASTA format
For instance, for HTT, we have decided to target a small repeat region spanning Chr4: 3,074,877-3,074,934 in the GRCh38.p11 as the ROI. To be certain of being able to design probes against this region, we downloaded a 10 kb chunk spanning chr4:3,070,000-3,080,000. Advanced users can perform this step by extracting the FASTA from an indexed reference using samtools faidx.
To maximize the probability that suitable probes will be found, we recommend limiting the probes to highly conserved regions wherever possible.
-
Search for crRNA probe sequences using CHOPCHOP.
Note: CHOPCHOP will only allow for a search within 20 kb at a time. For loci >20 kb, multiple searches should be performed, but the results from multiple searches can be pooled. It is possible to excise regions >20 kb using the Cas9 method, however the throughput will be significantly lower than for shorter regions. Generally, for regions >20 kb we recommend a tiling approach.
- Open CHOPCHOP in a web browser.
- Make sure the appropriate organism (in this case, H. sapiens) is selected in the central ‘In’ dropdown box. Regardless of the input sequence, the search for off-targets is performed against the selected organism. For example, to search for an E. coli sequence in a human background, where the human DNA is more abundant, H. sapiens should still be selected.
- Select ‘CRISPR/Cas9’ for Cas9 from the ‘Using’ dropdown box.
- To search within a FASTA sequence instead of genomic coordinates, click select ‘FASTA target’ and paste the FASTA sequence (<20 kb) in the box; otherwise, input genomic coordinates spanning a region <20 kb. Coordinates should take the format chrC:start-end, where ‘C’ is the chromosome, and ‘start’ and ‘end’ are the start and end coordinates, respectively (not containing comma separators).
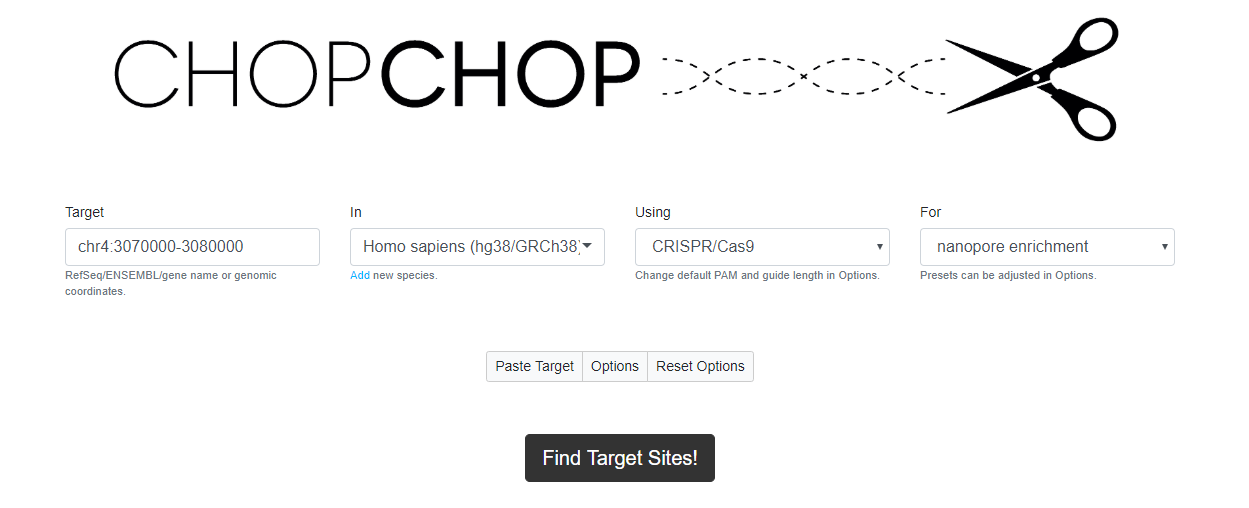
Figure 12. Initialising a Cas9 search using CHOPCHOP with specific genomic coordinates.
Tip: We do not recommend inputting a gene name as target, because for such searches CHOPCHOP currently only returns the results for exons.
- Select “nanopore-enrichment” under the ‘For’ dropdown to set the recommended preset. This will set the Efficiency score to be calculated using “Doench et al.2014 - only for NGG PAM”. We do not consider other scoring factors that are relevant only to genome editing in vivo.
- Click ‘Find Target Sites!’. The search may take a few minutes to complete.
- Download the results table by choosing “Results table” from the dropdown list next to “Download results”.
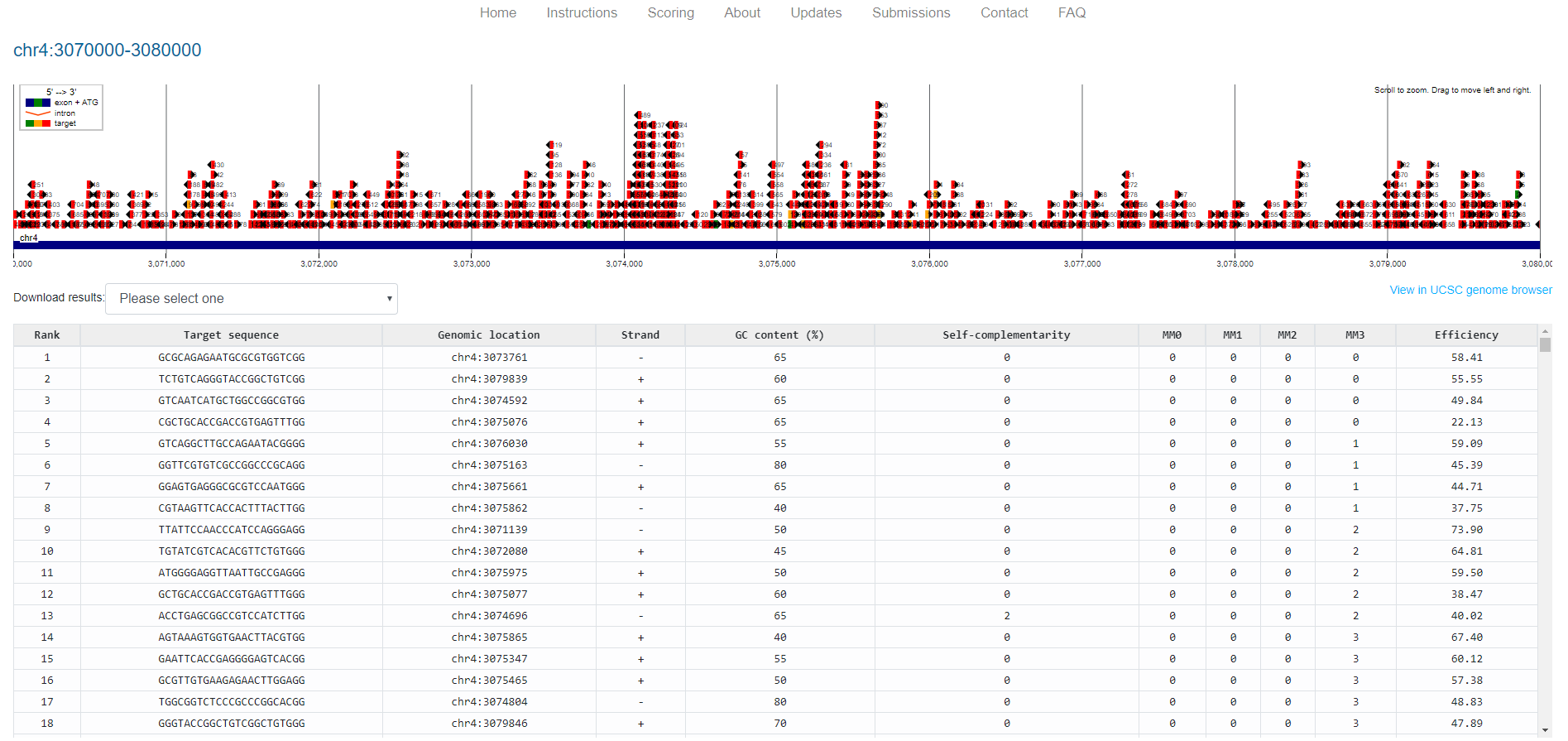
Figure 13. CHOPCHOP GUI, showing results for the input region in graphical and tabular format. The table can be downloaded for further filtering (see Step 3 below).
-
Filter the results table according to the following criteria:
- Retain GC between 40 and 80% for Cas9.
- Retain only crRNAs with a self-complementarity score of zero.
- Retain efficiency score > 0.3 (Cas9 only). This score predicts the efficiency of cutting based on the sequence rules (if “Doench et al., 2014” is selected above for “Efficiency score”).
- Retain candidates with the following mismatches: MM0 = 0; MM1 = 0; MM2 = 0; MM3 ≤ 5. Candidates with many predicted mismatches are likely to yield more off-target cuts. The severity of the off-target cut corresponds to the number of mismatches in the predicted sites: single-mismatch (MM1) sites are more likely to be cut by a probe than double (MM2) or triple (MM3) mismatches. Large numbers of mismatches should be avoided, especially with large >100-probe panels. In general, we recommend, where possible, filtering even more stringently for candidates with MM3 ≤ 2.
- Candidates should then be selected based on the desired cleavage location, the efficiency of the probe (higher is better), and the number of predicted mismatches.
If no probes are available in your target region, you may do the following:
- Expand the target region to include more flanking sequence;
- Slightly relax the filtering parameters, until sufficient candidates are found. We recommend relaxing the MM0 parameter to 1. -
Split the results table in two, separating the results for the (+) and (-) strand (Column ‘Strand’).
-
Retain the results for the (+) strand upstream of your target region (i.e., < Chr4: 3,074,877) and the results for the (–) strand downstream (i.e., > ChrX: 3,074,934).
-
Select two crRNAs for each of the (+) and (-) strands for redundancy.
-
Ensure that there are no SNPs in the probe and PAM regions, as this will reduce yield.
-
Order the crRNAs as Alt-R™ Cas9 crRNAs from IDT, following IDT’s online ordering instructions. The crRNA sequences should be provided as DNA sequences without PAMs using the instructions above.
The ordering page is presented here.
Note 1: The ordering page will convert the DNA sequence to RNA and will append any other relevant sequence automatically; this is required for the catalytic activity of Cas9.
Note 2: Please remember to order tracrRNA that will contain complementary regions to the crRNA - these are added by default in the IDT portal.
-
Examples of probes for HTT
Probe name HTT_2561 HTT_2662 HTT_7412 HTT_9569 gene HTT HTT HTT HTT sense + + - - xsome 4 4 4 4 allele maternal maternal maternal maternal location_NA12878 3072436 3072537 3077287 3079444 mm_in_seed FALSE FALSE FALSE FALSE GC 45 50 45 45 self_comp 0 0 0 0 MM0 1 1 1 1 MM1 0 0 0 0 MM2 0 0 0 0 MM3 1 2 2 1 efficiency 0.66 0.6 0.64 0.51 PAM AGG AGG AGG AGG The crRNA sequences for each of these probes are:
- HTT_2561 = TTTGCCCATTGGTTAGAAGC
- HTT_2662 = TCTTATGAGTCTGCCCACTG
- HTT_7412 = GGACAAAGTTAGGTACTCAG
- HTT_9569 = CTAGACTCTTAACTCGCTTG
These can be ordered from IDT and used in the initial step of the sample prep protocol as a control experiment or as an in-run control with other targets.

