-
The importance of good quality library
To make the most of a flow cell, load the amount of recommended good quality library recommended in the appropriate protocol for your sequencing kit. A good quality library is made up of sample molecules that have sequencing adapters ligated at both ends. To achieve high sequencing output, the flow cell membrane must not be damaged from bubbles and pore blocking should be at a minimum. There must also be enough library input to ensure the pores are always sequencing with minimal idle time between sequencing strands. In this section, we discuss how to achieve this.
How much to load on a flow cell is dependent on library preparation protocols, starting input and fragment length. For example, for the standard library preparation using the Ligation Sequencing Kit, we recommend a starting input of ~100-200 fmols for short fragments of <10 kb, or 1 µg of longer fragments of >10 kb. Starting input will also differ depending on user needs and starting input quality. For example, to generate ultra-long reads, ultra-long fragments must be present in the starting input sample.
It is important to load the recommended amount of library because this will affect data output. For example, not adding enough library onto the flow cell will impact pore occupancy and the optimal number of pores will not be sequencing DNA/RNA strands, reducing sequencing output.
Please refer to the Making the most of your flow cell for further information on how to interpret and use real-time data to improve sequencing output.
-
Quantifying DNA sample mass input for library preparation
To quantify the mass of DNA samples, we recommend the Qubit dsDNA BR Assay Kit. Both mass and length measurements are needed for a molar quantification of short-fragment samples.
To assess length, we recommend gel-based analysis or the Agilent 2100 Bioanalyzer. While molar quantification isn’t required for samples that comprise of long fragments, we still recommend measuring the length of your sample. This is to ensure your sample contains long fragments if you are interested in generating long reads. For this, we recommend using the pulsed-field gel electrophoresis or the Agilent Femto Pulse (the Agilent 2100 Bioanalyzer is not suitable for measuring the length of molecules >10 kb).
However, these are expensive pieces of equipment and are not accessible to every laboratory. Instead, it is possible to effectively QC the fragment length by running a small fraction of the library on a Flongle Flow Cell and evaluating the performance before deciding whether or not to commit to running the remainder of the library on a more expensive flow cell.
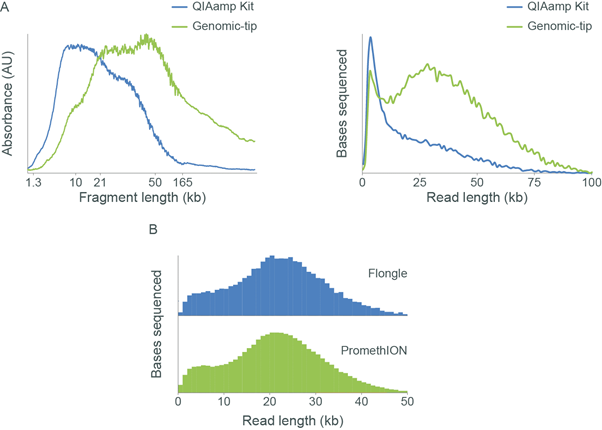
Assessing fragment length of DNA samples using the Agilent Femto Pulse and the Oxford Nanopore Technologies Flongle Flow Cell. Panel A: Genomic DNA was extracted from rabbit blood (collected in K2-EDTA) using either the QIAamp DNA Blood Midi Kit or a QIAGEN Genomic-tip. The extracted gDNA was analysed using the Agilent Femto Pulse (left). Libraries were prepared using the Ligation Sequencing Kit and sequenced on a MinION Flow Cell, and the read length distributions are shown (right). The Femto Pulse trace shows that the QIAamp extracted sample consists of mainly short fragments (<10 kb) and this is reflected in the sequencing. Whereas the Genomic-tip extracted sample is shown to contain a higher proportion of longer fragments and therefore, produces a higher proportion of longer reads. Panel B: Genomic DNA was extracted from GM12878 cells using the QIAGEN Gentra Puregene Cell Kit and a sequencing library was prepared using the Ligation Sequencing Kit. A portion of the sequencing library was run on a Flongle Flow Cell and the remainder on PromethION Flow Cell. The observed read length distributions are reproducible between the platforms. -
Purity of DNA samples
It is also recommended to measure the purity of extracted DNA. DNA extraction methods often involve chemicals that can inhibit enzymatic activity, including those enzymes in library preparation.
To measure the presence of certain contaminants, the Nanodrop 2000 Spectrophotometer can be used. Pure DNA gives an A260/A280 ratio of ~1.8. A ratio lower than this can be indicative of a contaminant such as phenol. A A260/A230 ratio lower than 2.0–2.2 can also be indicative of phenol contamination but can also indicate carryover of guanidine or carbohydrate. However, while Nanodrop readings are a good place to start, it is not the only authority on purity and subsequent performance. Details of the effects of certain contaminants on the efficiency of the Ligation Sequencing Kit can be found in the DNA contaminants and RNA contaminants documents.
If you have low starting amounts of DNA (<100 ng), PCR is recommended to generate more template. There are library preparation kits and protocols available specifically for PCR-based preparation:
Ligation sequencing V14 - low input by PCR (SQK-LSK114 with EXP-PCA001)
Rapid sequencing DNA - PCR Barcoding Kit 24 V14 (SQK-RPB114.24)
The choice of kit is dependent on the priorities of the user.
If inputs below the protocol recommendations are used, the sequencing output may be negatively affected due to the low pore occupancy.
To maximise sequencing output, it is important that the pores are kept filled with DNA to minimise the time that they are “idle” in-between strands. This metric can be monitored by viewing the pore occupancy graph on the MinKNOW UI. For more information, see the MinKNOW protocol.
-
Pore occupancy and library quality
A good quality library is also required and refers to the extent of how well the DNA has been adapted with sequencing adapters. Molecules without sequencing adapters cannot be sequenced and diminish library quality. Libraries that consist of molecules with adapters and tethers on both ends are more potent than those with only one end modified. Note: Equivalent library loads may carry slightly different potency (in terms of pore occupancy) between Flongle, MinION and PromethION Flow Cells which is reflective of the differences in flow cell volume (37.5 µl for Flonge, 75 µl from MinION and 200 µl for PromethION).
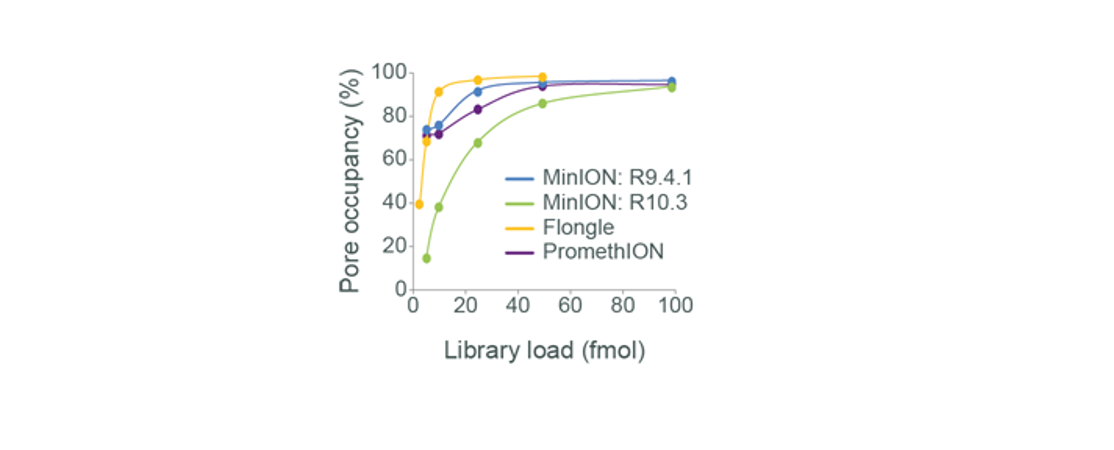
The relationship between the amount of library loaded onto an Oxford Nanopore Technologies flow cell and the resulting pore occupancy that was obtained. We found that pore occupancy is maximised when ~5–50 fmol of good quality library is loaded onto R9.4.1 flow cells and ~25–75 fmol is loaded onto R10.3 flow cells.The less library you load, the fewer “threadable ends” will be present to be captured by the pores. Therefore, the pores will be “searching” for molecules for longer, and if the pores are not always sequencing, then output could be compromised. It is important to note that we do not observe a linear relationship between input onto the flow cell and sequencing yield, but loading less could give reduced output. Conversely, loading more library does not guarantee good performance.
-
Determinants of read length
We have found that long reads can be achieved with both ligation- and rapid-based library preparation methods, as read length reflects input fragment length and is not platform-limited. For example, fragmenting input DNA results in a narrower distribution of read lengths. In some kits, we recommend fragmenting input DNA to ease handling and quantification or to make multiple samples a uniform size. We have also validated several methods of enrichment for long reads, including using SPRI beads and the Short Fragment Eliminator Kit (EXP-SFE001) that are located in the linked Size Selection folder.
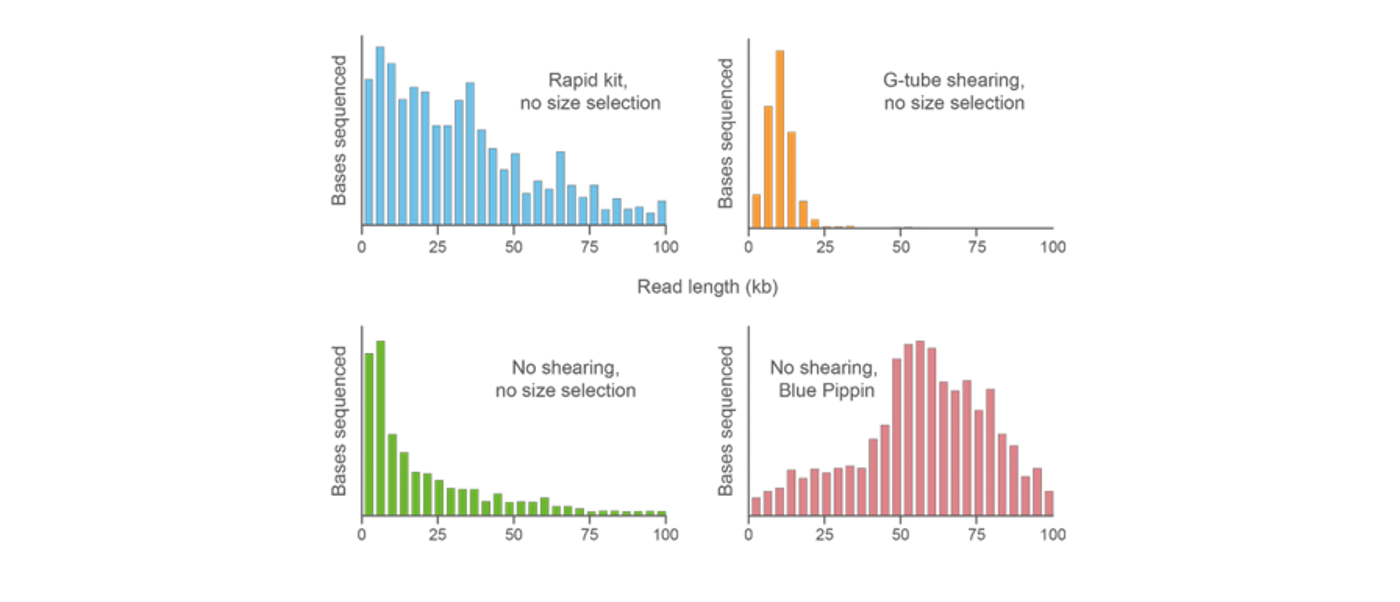
A DNA library was prepared using the Rapid Sequencing Kit. Top left: Read length from the sequencing kit. Top right: Read length with only g-TUBE shearing. Bottom left: No fragmentation or size selection. Bottom right: Read length using BluePippin for size selection. -
Size selection
Size selection is an optional step to enrich longer fragments in library input. SPRI-bead purification is used in many Oxford Nanopore Technologies library preparation protocols as this technique is effective at removing fragments of 1.5 kb and can slightly shift read length towards the longer end. The Short Fragment Eliminator Kit (EXP-SFE001) may be used to remove fragments of <25 kb and almost completely removing fragments under 10 kb. However, both methods effectively remove shorter fragments and boost read N50 without compromising data output. To view our protocols for size selection, please refer to the linked Size selection page.
-
Fragment length
The pores in a flow cell can sequence all sizes of fragments and read lengths are only limited by template molecule starting length. Nanopores capture and process both long and short DNA and RNA strands. However, the length of fragments can affect output as the concentration of free DNA ends will be different. For example, short fragments have a higher concentration of DNA ends that will increase pore occupancy and output. This is because there are more threadable ends for the pores to capture.
Libraries composed of long fragments typically have lower data outputs than short fragments as there are fewer molecules available for sequencing and they take longer to be captured by the pore. This also results in more pores not sequencing due to the lack of pore occupancy. If size selection is performed as part of the library preparation, this can lower output further due to fragments outside a particular range being excluded.
Fragmentation in a Covaris g-TUBE has been found to yield the highest output with no size selection. Therefore, we recommend using this fragmentation method in many protocols to optimise for highest output yield.
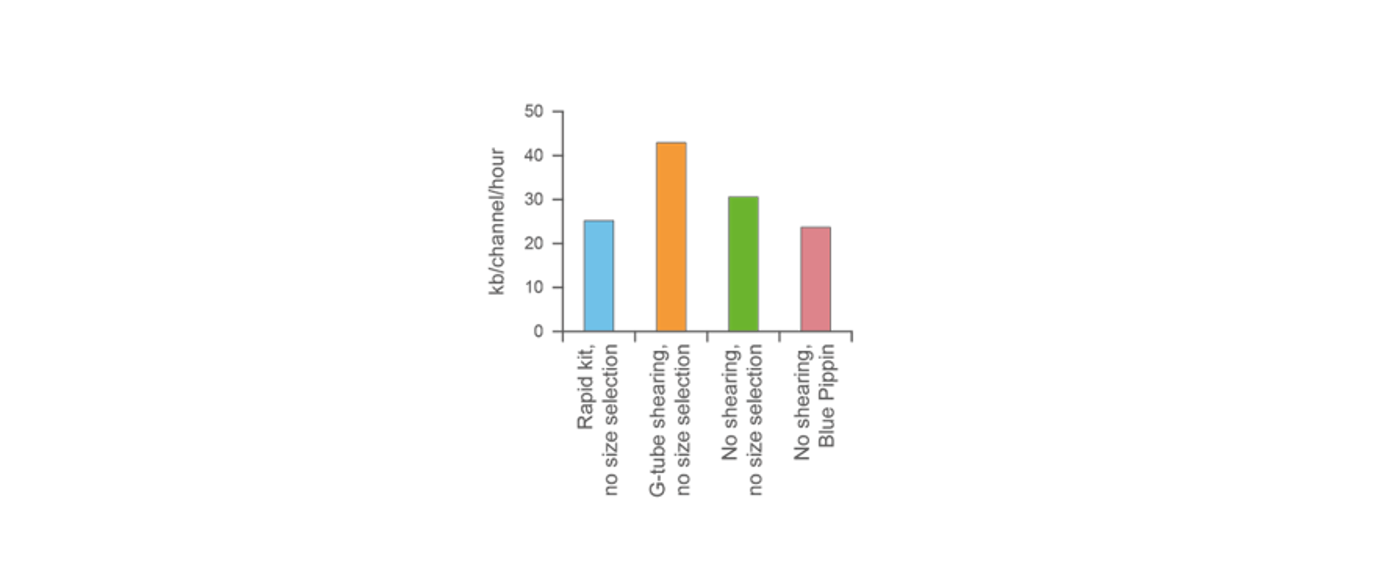
G-tube fragmentation yields the highest output. -
Flow cell performance
Flow cell output is governed by various factors, including DNA/RNA library input, loading amounts, and pore blocking. Below, we review how controlling the input material size distribution by fragmentation impacts these factors and what Oxford Nanopore Technologies’ recommendations are to generate the best data, based on experimental aims.
Increasing read N50
It has been observed that some shearing of gDNA samples can lead to an increase in observed read length: this seems counterintuitive – how can breaking up the DNA fragments give longer reads? It has been suggested that certain fragments may be so long that they become “lost” during the library preparation and therefore are not observed, leaving only the short fragments (for example, the very longest fragments may not efficiently bound to, or elute from the AMPure beads used after end-prep or ligation). Light shearing, for example using the Megaruptor, can break up the very longest molecules into chunks that the library preparation can more readily process, leading to increased read N50s.
This approach is suggested for users where samples appear to be very high molecular weight in gel or Femto Pulse analysis but the observed read length N50 is <15 kb. Other users within the Nanopore Community have also attempted other shearing methods to increase read lengths. However, the more aggressive the fragmentation, the higher the risk of over-fragmenting, leading to a reduction in observed read lengths.
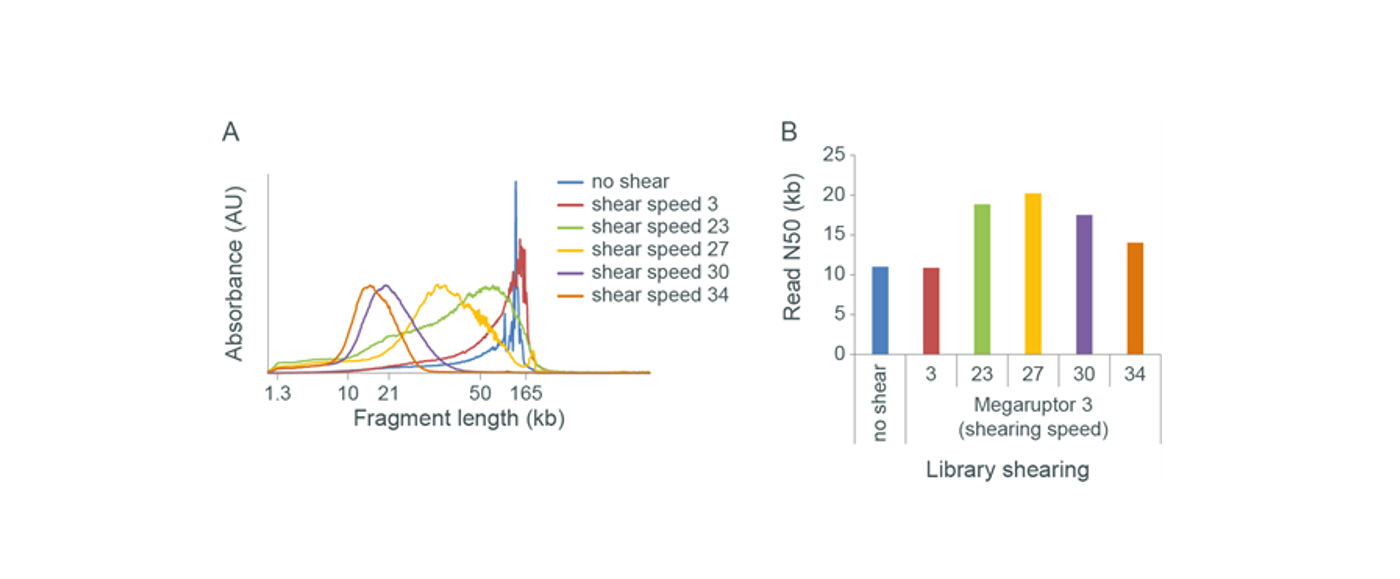
The effect of shearing very high molecular weight gDNA with Megaruptor 3. Human gDNA was extracted from cell culture, with the aim to recover the longest possible fragments. The resulting gDNA was sheared with Megaruptor 3 using a selection of shearing speeds. Panel A: The sheared DNA was analysed by Femto Pulse. The sequencing libraries were prepared using the Ligation Sequencing Kit and run on a MinION Flow Cell. Panel B: The read N50 values were recorded. The read length distribution of the input (no shear) shows that most of the DNA is above 100 kb, with a spike at 165 kb (area where fragments become compressed). However, this does not correspond to a high read N50 in sequencing. The lowest shearing speed had little-to-no effect on the fragment length distribution or the observed read N50, suggesting unsuccessful fragmentation. However, increasing the shearing speed to 20–30 did show successful fragmentation and led to an increase in observed read N50. Increasing the shearing speed still further led to over-fragmentation and a drop in observed read N50.Input amount and pore occupancy
Loading too much or too little library can compromise flow cell performance. For example, 5–50 fmol is optimal for our previously available R9.4.1 flow cells. If sufficient starting material is unavailable, users can start with lower inputs, however we have found that data output can drop at lower inputs as there are insufficient molecules available to maximise pore occupancy. Fragmenting the sample (for example using a Covaris g-TUBE or Megaruptor®) can be used to increase the number of molecules/ends to thread into the nanopores. This increases pore occupancy and recovers the output: an input of 100 ng unsheared Lambda DNA results in a flow cell load of ~1 fmol, which can be increased to ~6 fmol by shearing with a g-TUBE.
It is worth noting that fragmenting DNA to boost the output can mean it may not be possible to achieve ultra-long reads. If you have <100 ng of DNA, we advise performing PCR to increase the amount of DNA available for sequencing.
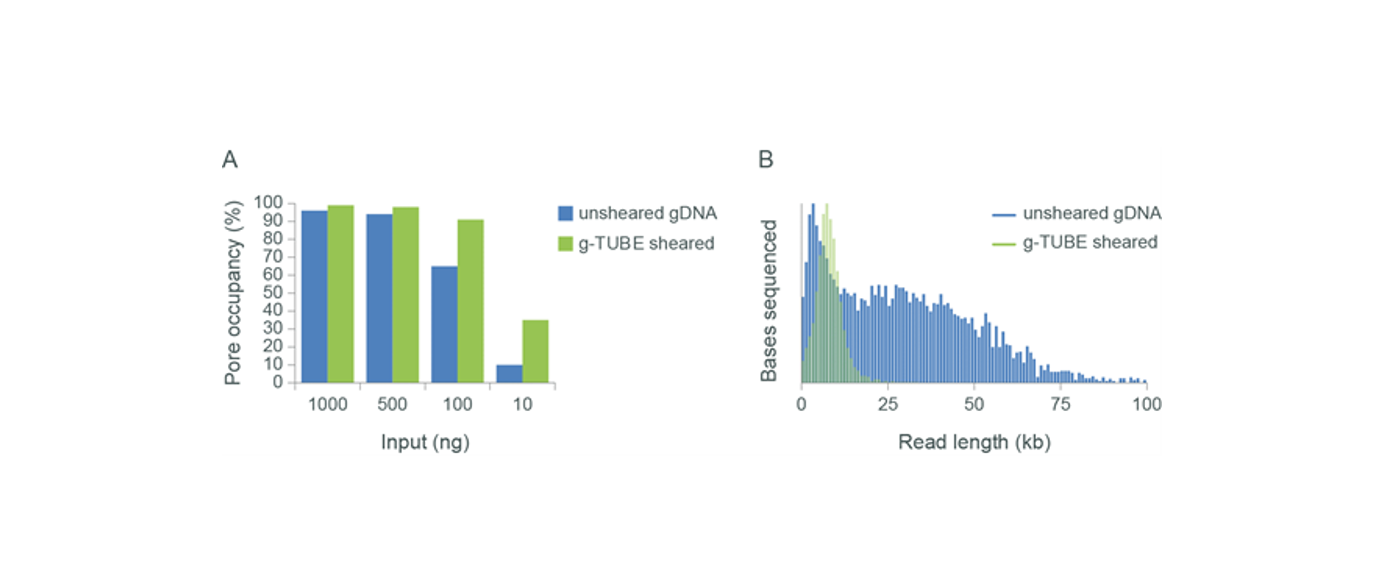
The relationship between input and output (R9.4.1 flow cells) for sheared and unsheared libraries using our previous sequencing chemistry. Panel A: As the input of unsheared gDNA into the library preparation drops below ~500 ng, the pore occupancy decreases, leading to a decrease in flow cell output (Gbases). Shearing the sample using a Covaris g-TUBE increases the molar concentration of the sample leading to more efficient use of the pores and an increase in flow cell output. Panel B: Shearing samples with a g-TUBE impacts the read length distribution.We investigated the performance that can be obtained from human gDNA when starting with 100 ng, with and without shearing, using our previously available R9.4.1 flow cells using MinION and PromethION. It was observed that even without shearing, ~10 Gbases and ~30 Gbases could be obtained from as little as 100 ng of HMW gDNA. Pore occupancy and flow cell output was increased when the sample was sheared with both Megaruptor® 3 and Covaris g-TUBE.
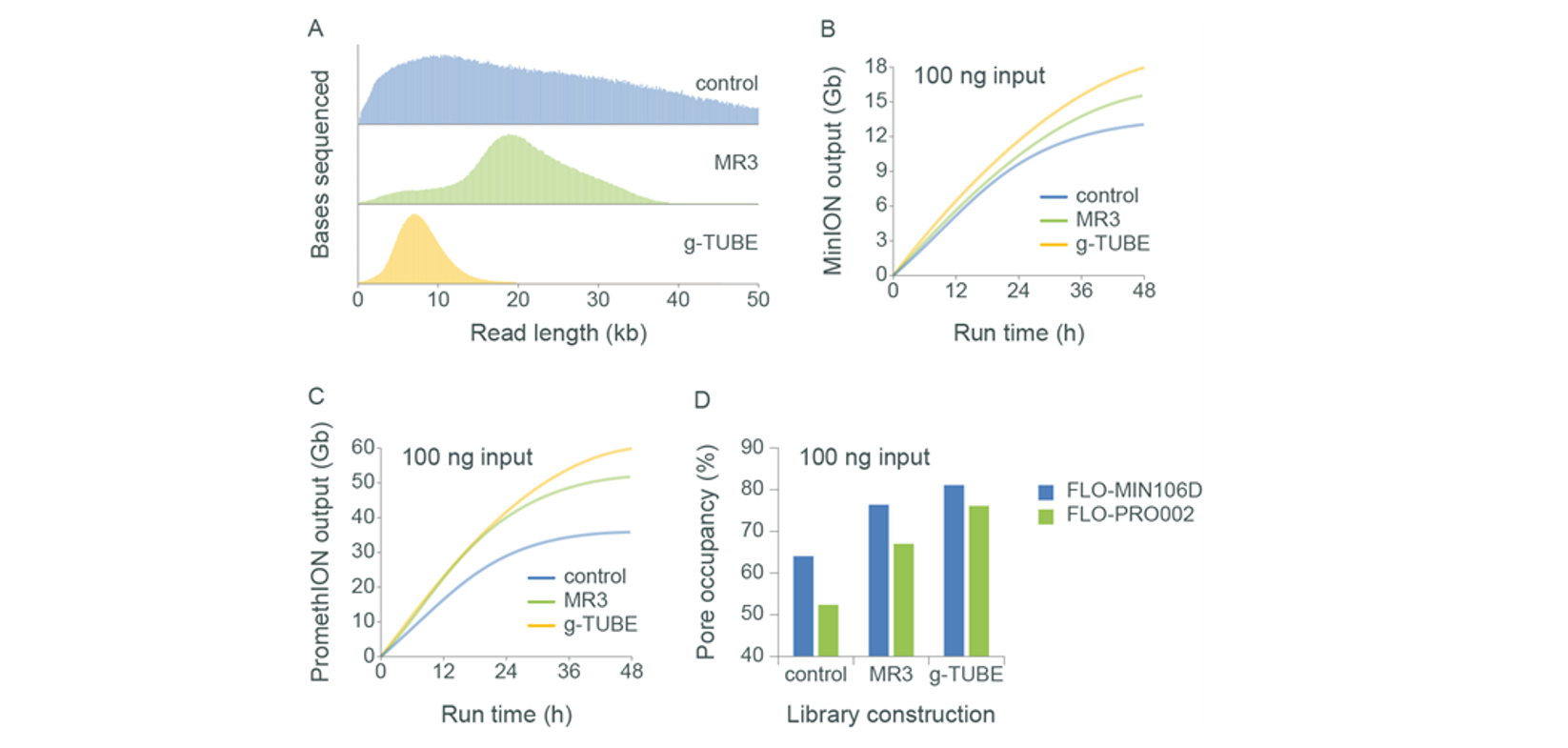
Output on MinION and PromethION Flow Cells with 100 ng of HWM human gDNA. Human gDNA was extracted from GM24385 cells using the QIAGEN Gentra Puregene Cell Kit. The HMW gDNA was sheared using either Megaruptor® 3 (speed setting 30) or Covaris g-TUBE. Libraries were prepared for sequencing with the Ligation Sequencing Kit using 100 ng of sheared and unsheared template DNA. The libraries were run on R9.4.1 flow cells using MinION and PromethION. Panel A: Shearing the input gDNA decreases the read lengths that are observed in the subsequent sequencing. Panel B and C: The flow cell output (Gbases) obtained from 100 ng of input is increased by shearing on both MinION and PromethION, respectively. Panel D: The increased shearing of the sample increases the pore occupancy efficiency, leading to the higher outputs from the sheared samples. At low inputs (100 ng of gDNA) the pore occupancy is observed to be higher on MinION compared to PromethION.Blocking
Pore blocking is another factor that can affect flow cell output. During a sequencing run, pores can become “blocked”, preventing the pore from accepting a new strand for sequencing or continuing to sequence the occupying strand. Such blocks are detected by the MinKNOW software, which changes the channel state from “single pore” to “unavailable”. For the duration of a blockage, the pore acquires no sequencing data. MinKNOW attempts to drive out whatever has blocked the pore by reversing the voltage. The unblocking scheme is progressive, increasing the duration of the voltage reversal until the blockage is cleared. Most of the time (~98%), attempts to unblock a pore are successful and it reverts to the single pore state, where it is available to accept new strands and continue sequencing. However, in a minority of cases, the progressive unblocking scheme will not be able to recover a blocked pore. If a pore becomes terminally blocked and cannot be recovered, a new pore is swapped in from a different well in the channel, if available. Typically, a blockade occurs every 250–500 kb and is successfully removed ~98% of the time (in other words, around 1 in 50 attempts will be unsuccessful). This gives an average output of ~10–20 Mbases per pore, which for a flow cell containing ~1500 pores could lead to a total output of ~15–30 Gbases (note, other factors may limit the actual output obtained). If there is an increase in the rate of blocking, then pores spend less time sequencing and more unblocks are triggered. If there are more unblocks, or if the success of unblocking decreases, then the rate at which pores are lost increases and total flow cell output is reduced.
To determine if fragment length played a role in the rate of blocking, we took DNA extracted from human cells grown in culture (GM12878) and sheared it with Megaruptor 2. We were not able to establish a relationship between read length and blocking rate, although we observed a decrease in the success rate of the unblock for the longer libraries, indicating that our unblocking scheme is less capable of removing blocks from longer fragments. Given this observation, if users are obtaining a low output, then some shearing of the sample could be performed to see if unblocking success can be improved to help boost output.
Recommendations for libraries with high levels of blocking:
- Fragment the starting DNA to increase pore occupancy
- Flow cell washing may be used to unblock pores and revert ‘unavailable’ state to ‘single pore’ state. The nuclease in the Flow Cell Wash Kit is able to digest all of the sample remaining on the flow cell and contaminants blocking pores to increase output for more library to be loaded and sequenced.
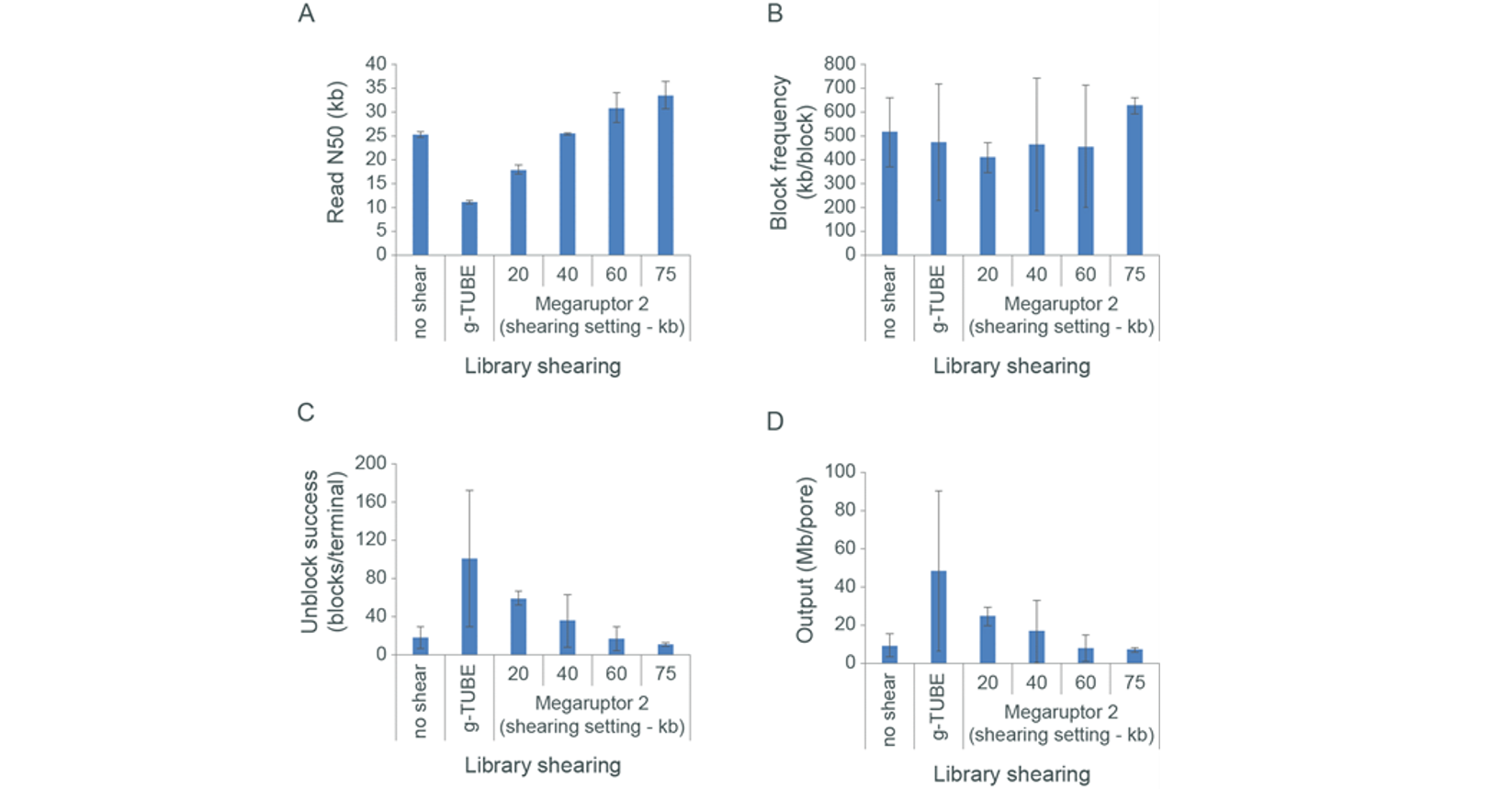
The effect of read length on blocking, unblocking and flow cell output. Extracted gDNA was sheared with Megaruptor 2 using different shearing settings, or with a Covaris g-TUBE. Libraries were then prepared using the Ligation Sequencing Kit and sequenced on the MinION. Panel A: The read N50 values for the differently sheared libraries. Note, some Megaruptor shearing produces a slightly elevated read N50. Panel B: The block frequency (kb/block) for the differently sheared libraries suggests that there is little relationship between the frequency at which blockades occur and the length of the fragments that are being sequenced, at least for this sample. Panel C: As the fragment length of the library increased, a decrease in rate of the success of the unblock (number of blocks before a terminal block was encountered) was observed. Panel D: As a result of the decrease in unblock success with increased read lengths, it follows that the output from any pore may also decrease with increased read length.Optional size selection
Users of Oxford Nanopore Technologies platforms have observed reads of >1 Mbases and the longest read observed approaches 2.3 Mbases. To obtain long reads, long fragments must be present in the extracted DNA sample. All extraction techniques that we recommend yield some short fragments (<10 kb) and even if they do not appear prevalent during quantification, it is likely that you will observe some short reads. Size selection can be used to enrich for long fragments or deplete short fragments. Our size selection protocols are available on the size selection page.
The SPRI bead method enriches for molecules above ~1.5 – 2 kb and the size selection buffer (SSB) promotes semi-selective precipitation and enrichment of molecules above ~10 kb. The BluePippin has a tuneable cut-off limit up to 40 kb. To demonstrate the performance, gDNA was size selected using each method; the BluePippin instrument settings were set to enrich for molecules >40 kb. Using the Ligation Sequencing Kit, 1 µg of each size-selected DNA was prepared for sequencing and the libraries were run on the MinION. A library where no size selection had been performed was also sequenced as control. The data below shows the read length distributions observed and the output generated for each of the libraries. Very little difference in output was observed over the course of the 24 hour sequencing run for SPRI and SSB size-selected libraries, compared with the control. However, an accumulation of pores in the unavailable state were observed throughout the course of the run for the BluePippin size selected library, so a flow cell wash was performed after ~16 hours and more library loaded before recommencing the run.

Size selection of gDNA samples. Genomic DNA was size selected and then prepared for sequencing using the Ligation Sequencing Kit. The read N50 values of the control, SPRI bead size-selected, our Short Fragment Eliminator (EXP-SFE001) size-selected, and BluePippin size-selected libraries were ~20 kb, ~25 kb, ~35kb and ~41 kb, respectively. Very little difference in output was observed over the course of the 24 hour sequencing run for SPRI and EXP-SFE001 size-selected libraries, compared with the control. However, an accumulation of pores in the unavailable state was observed for the BluePippin size selected library, so a flow cell wash were performed after ~16 hours and more library loaded before continuing the run.








